BIN_ASSESSMENT#
 This module is a part of metaFun pipeline, designed for assessing genome quality and assigning taxonomy to metagenome-assembled genomes (MAGs).
This module is a part of metaFun pipeline, designed for assessing genome quality and assigning taxonomy to metagenome-assembled genomes (MAGs).
Overview#
The BIN_ASSESSMENT module is the third step in the metaFun pipeline, designed for comprehensive evaluation of metagenome-assembled genomes (MAGs). It performs quality assessment using CheckM2, contamination assessment using GUNC, taxonomic classification using GTDB-Tk, and combines the results for downstream analysis. This module helps identify high-quality genomes suitable for comparative genomic analyses.
Module Execution#
# Basic usage
(metafun) metafun -module BIN_ASSESSMENT
# Specify input directory if you used a custom output path in ASSEMBLY_BINNING
(metafun) metafun -module BIN_ASSESSMENT -i /path/to/bins
# Provide metadata file for combining with quality/taxonomy results (recommended)
(metafun) metafun -module BIN_ASSESSMENT -m your_metadata.csv -c 2
# Specify a quality filter for selecting genomes
(metafun) metafun -module BIN_ASSESSMENT --pass_quality medium_quality.pass
Quality filtering options
There are several options for filtering genomes based on quality metrics:
For basic quality filtering, use
medium_quality.pass:
(metafun) metafun -module BIN_ASSESSMENT --pass_quality medium_quality.pass
For stricter quality requirements, use
high_quality.pass:
(metafun) metafun -module BIN_ASSESSMENT --pass_quality high_quality.pass
For including genomes that pass both quality and GUNC checks:
(metafun) metafun -module BIN_ASSESSMENT --pass_quality QS50_gunc.pass
Available quality filter options:
medium_quality.pass: Completeness ≥ 50%, Contamination < 10%high_quality.pass: Completeness > 90%, Contamination < 5%medium_quality_gunc.pass: medium_quality + GUNC passhigh_quality_gunc.pass: high_quality + GUNC passQS50.pass: QS50 score ≥ 50QS50_gunc.pass: QS50 pass + GUNC passall: No quality filtering (include all genomes)
Module Operation Sequence#
This module performs the following steps:
Preparation of input genome bins
Genome quality assessment using CheckM2
Genome contamination assessment using GUNC
Combining CheckM2 and GUNC results and filtering genomes based on quality
Taxonomic classification of quality-filtered genomes using GTDB-Tk
Creating a final report combining quality assessment and taxonomic classification
(Optional) Combining results with user-provided metadata
Parameters#
${launchDir} is the directory where you execute metaFun, and utilized as output base directory.
Parameter |
Description |
Default Value |
Note |
|---|---|---|---|
|
Input directory with genome bins |
|
Output from ASSEMBLY_BINNING module or specify your own directory containing genome bins. |
|
Output directory |
|
Default recommended for downstream analysis |
|
Path to metadata file |
null |
Optional but recommended. Used for combining quality/taxonomy results with sample metadata for downstream analysis. |
|
Column index in metadata |
1 |
Specifies which column in the metadata file contains the sample identifiers that match genome bin names. |
|
Quality filter for genome selection |
|
Options: |
|
Number of CPUs to use |
20 |
Adjust based on your system capabilities |
|
Unique run identifier |
Timestamp_workflowName |
Generated automatically in format “yyyyMMddHHmmss_number” (e.g., “20250306135829_2996”). Can be customized if needed. |
About parameters for quality filtering and metadata
Quality Filtering Implementation:
The Nextflow script implements the quality filtering based on the --pass_quality parameter. When genomes are processed:
The parameter selects which genomes will be passed to subsequent steps based on the specified quality criteria.
If
--pass_qualityis set toall, all genome bins are included without filtering.For other options (e.g.,
QS50.pass,medium_quality.pass), the script filters genomes based on the corresponding column in the combined quality report, including only those marked as “True” for the selected quality criterion.
Accession Column Example:
Consider this example of how --accession_column works with a metadata file:
Metadata file (sample_metadata.csv):
run_accession,sample_id,location,disease_status,age
SRR6915091,P001,USA,Healthy,35
SRR6915092,P002,Canada,Disease,42
SRR6915093,P003,UK,Healthy,28
Your genome bin names would be something like:
SRR6915091_bin.1.fa
SRR6915091_bin.2.fa
SRR6915092_bin.1.fa …
To match these bin names with the metadata, you would use:
metafun -module BIN_ASSESSMENT -m sample_metadata.csv -c 1
Because column 1 (run_accession) contains the sample identifiers (SRR6915091, SRR6915092) that match the prefix of your genome bin names.
Inputs and Outputs#
Inputs#
Genome bins in FASTA format (
.fa,.fna, or.fastaextensions)These should be the high-quality bins from the ASSEMBLY_BINNING workflow
Default input directory:
${launchDir}/results/metagenome/ASSEMBLY_BINNING/final_binsOptional: Metadata file in CSV or TSV format with sample information
Outputs#
CheckM2 quality assessment results
GUNC contamination assessment results
GTDB-Tk taxonomic classification
Quality-filtered genome bins
Combined quality and taxonomy report
Optional: Combined metadata with quality/taxonomy information
Output directory structure#
Output directory is at ${launchDir}/results/metagenome/BIN_ASSESSMENT or your specified directory path with -o outdir.
Switching input and output directory.
If you define a custom output directory with -o ${output directory}, you should modify input parameters in downstream workflows accordingly.
The default output directory is results/metagenome/BIN_ASSESSMENT in your ${launchDir}.
${launchDir}/results/metagenome/BIN_ASSESSMENT/
├── prepared_bins_${run_id}/ # Prepared and renamed input bins
│ └── renamed_bins/
│ ├── ${sample_id}_bin.1.fa
│ ├── ${sample_id}_bin.2.fa
│ └── ...
├── checkm2_${run_id}/ # CheckM2 quality assessment results
│ └── ${outputDirCheckM2}/
│ ├── quality_report.tsv # Main quality assessment report
│ ├── protein_files/ # Predicted proteins from genomes
│ │ ├── ${genome_name}.faa
│ │ └── ...
│ └── ...
├── gunc_${run_id}/ # GUNC contamination assessment results
│ └── ${outputDirGUNC}/
│ ├── GUNC.progenomes_2.1.maxCSS_level.tsv # Main contamination report
│ └── ...
├── checkm_gunc_combined_${run_id}/ # Combined quality/contamination results
│ └── combined_report.tsv # Combined CheckM2 and GUNC results
├── bins_quality_passed/ # Quality-filtered genome bins
│ ├── ${sample_id}_bin.1.fa # Only bins passing quality filter
│ └── ...
├── gtdb_outdir_${run_id}/ # GTDB-Tk taxonomy classification results
│ ├── gtdbtk.bac120.summary.tsv # Bacterial genome classifications
│ ├── gtdbtk.ar53.summary.tsv # Archaeal genome classifications
│ ├── quality_taxonomy_combined.csv # Combined quality and taxonomy
│ └── ...
└── quality_taxonomy_combined_final.csv # Final report for downstream analysis
Additionally, when metadata is provided:
${launchDir}/
├── combined_metadata_quality_taxonomy_${run_id}.csv # Combined metadata and quality/taxonomy
└── metadata_column_BIN_ASSESSMENT_summary.tsv # Summary of metadata columns
Execution Examples and Results#
metaFun command line execution example#
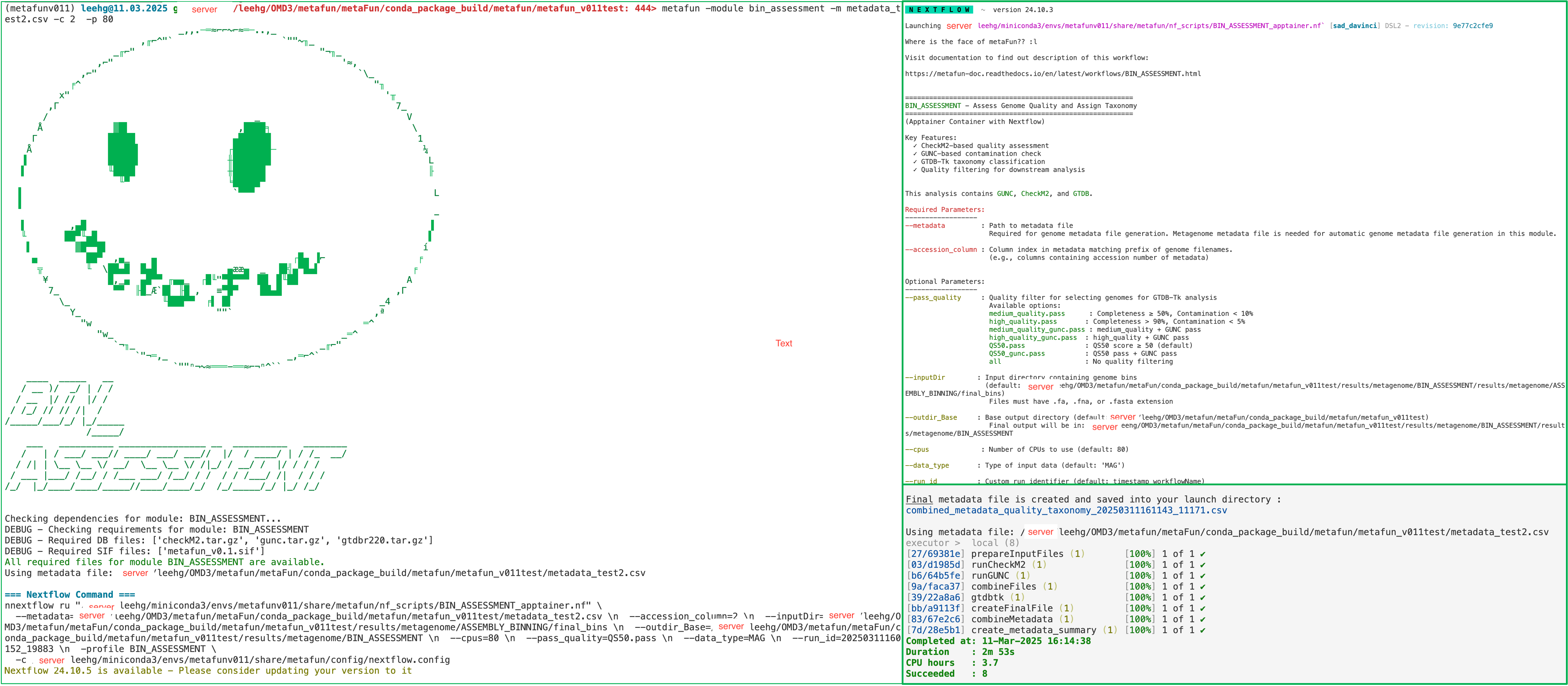
Combining metadata with quality/taxonomy results
The combined metadata file (combined_metadata_quality_taxonomy_${run_id}.csv) is essential for downstream analysis in the GENOME_SELECTOR module. It links genome quality and taxonomy with sample metadata. The bins selected in this process will be used for further analysis in the COMPARATIVE_ANNOTATION module.
To work effectively with your metadata files:
First, check the metadata column summary to identify the column structure:
cat yourmetadata.csv
You need to identify accession column for you metadata
This helps you understand your data’s structure before proceeding to genome selection and further analysis.
Example of Quality Assessment Results#
$ head -n 5 ${launchDir}/results/metagenome/BIN_ASSESSMENT/checkm2_*/quality_report.tsv
Name Completeness Contamination Completeness_Model_Used Translation_Table_Used Coding_Density Contig_N50 Average_Gene_Length Genome_Size GC_Content Total_Coding_Sequences Total_Contigs Max_Contig_Length Additional_Notes
SRR6915091_bin.1 98.2 1.3 Neural Network (Specific Model) 11 0.877 278453 899.8 2758979 0.474 2698 823 32542 None
SRR6915091_bin.2 87.6 3.2 Neural Network (Specific Model) 11 0.863 129876 891.2 3104325 0.418 3077 945 28754 None
SRR6915091_bin.3 62.4 5.1 Gradient Boost (General Model) 11 0.901 87542 911.5 2198765 0.562 2154 752 18923 None
The CheckM2 quality report includes the following key columns:
Name: Genome bin identifier
Completeness: Estimated genome completeness percentage (0-100%)
Contamination: Estimated contamination percentage (0-100%)
Completeness_Model_Used: Model used for estimating completeness (e.g., “Neural Network (Specific Model)”, “Gradient Boost (General Model)”)
Translation_Table_Used: Genetic code used for translation
Coding_Density: Proportion of the genome that codes for proteins
Contig_N50: N50 value of contigs (length where 50% of the genome is in contigs of this size or larger)
Average_Gene_Length: Average length of predicted genes
Genome_Size: Total size of the genome in base pairs
GC_Content: GC content of the genome (0-1)
Total_Coding_Sequences: Number of predicted coding sequences
Total_Contigs: Number of contigs in the genome assembly
Max_Contig_Length: Length of the largest contig
Example of Taxonomy Classification Results#
$ head -n 5 ${launchDir}/results/metagenome/BIN_ASSESSMENT/gtdb_outdir_*/gtdbtk.bac120.summary.tsv
| Genome | Completeness | Contamination | medium_quality.pass | near_complete.pass | medium_quality_gunc.pass | near_complete_gunc.pass | QS50 | QS50.pass | pass.GUNC | QS50_gunc.pass | classification | Analysis_accession | bioproject_accession | accession_used_in_analysis | country | continent | host_age | host_body_mass_index | host_sex | disease_group | AJCC_stage | age_group |
|-----------------------|--------------|---------------|---------------------|--------------------|--------------------------|-------------------------|-------|-----------|-----------|----------------|------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------|--------------------|----------------------|----------------------------|---------|-----------|----------|----------------------|----------|---------------|------------|-----------|
| SRR6915091_MB2.14 | 91.77 | 0.55 | True | True | True | True | 89.02 | True | True | True | d__Bacteria;p__Bacillota_C;c__Negativicutes;o__Acidaminococcales;f__Acidaminococcaceae;g__Acidaminococcus;s__Acidaminococcus intestini | SRR6915091 | PRJNA447983 | SRR6915091 | Italy | Europe | 77.0 | 23.0 | Male | CRC | | Old |
| SRR6915091_MB2.16 | 93.32 | 6.58 | True | False | False | False | 60.42 | True | False | False | d__Bacteria;p__Bacteroidota;c__Bacteroidia;o__Bacteroidales;f__Bacteroidaceae;g__Phocaeicola;s__Phocaeicola dorei | SRR6915091 | PRJNA447983 | SRR6915091 | Italy | Europe | 77.0 | 23.0 | Male | CRC | | Old |
| SRR6915091_MB2.17 | 70.82 | 2.2 | True | False | True | False | 59.82 | True | True | True | d__Bacteria;p__Bacillota_A;c__Clostridia;o__Lachnospirales;f__Lachnospiraceae;g__Fusicatenibacter;s__Fusicatenibacter saccharivorans | SRR6915091 | PRJNA447983 | SRR6915091 | Italy | Europe | 77.0 | 23.0 | Male | CRC | | Old |
| SRR6915091_MB2.18_sub | 64.01 | 9.25 | True | False | False | False | 17.76 | True | False | False | d__Bacteria;p__Bacillota_A;c__Clostridia;o__Oscillospirales;f__Oscillospiraceae;g__Flavonifractor;s__Flavonifractor plautii | SRR6915091 | PRJNA447983 | SRR6915091 | Italy | Europe | 77.0 | 23.0 | Male | CRC | | Old |
```{code-block} bash
:caption: Example of combined quality and taxonomy report
$ head -n 5 ${launchDir}/results/metagenome/BIN_ASSESSMENT/quality_taxonomy_combined_final.csv
Genome,Completeness,Contamination,medium_quality.pass,high_quality.pass,medium_quality_gunc.pass,high_quality_gunc.pass,QS50,QS50.pass,pass.GUNC,QS50_gunc.pass,classification
SRR6915091_bin.1,98.2,1.3,True,True,True,True,89.02,True,True,True,d__Bacteria;p__Bacteroidota;c__Bacteroidia;o__Bacteroidales;f__Bacteroidaceae;g__Bacteroides;s__Bacteroides vulgatus
SRR6915091_bin.2,87.6,3.2,True,False,True,False,72.76,True,True,True,d__Bacteria;p__Firmicutes_A;c__Clostridia;o__Lachnospirales;f__Lachnospiraceae;g__Blautia;s__Blautia obeum
SRR6915091_bin.3,62.4,5.1,True,False,False,False,47.05,False,False,False,d__Bacteria;p__Firmicutes_A;c__Clostridia;o__Oscillospirales;f__Ruminococcaceae;g__Faecalibacterium;s__Faecalibacterium prausnitzii
Nextflow Processes in BIN_ASSESSMENT Module#
Process |
InputDir |
OutputDir |
Note |
|---|---|---|---|
prepareInputFiles |
|
|
Prepares and renames input genome bins |
runCheckM2 |
Output from prepareInputFiles |
|
CheckM2 quality assessment |
runGUNC |
Output from runCheckM2 |
|
GUNC contamination assessment |
combineFiles |
Outputs from runCheckM2 and runGUNC |
|
Combines results and filters genomes |
gtdbtk |
Outputs from combineFiles |
|
GTDB-Tk taxonomic classification |
createFinalFile |
Outputs from gtdbtk |
|
Creates final combined report |
combineMetadata |
Final report and user metadata |
|
Optional: Combines with user metadata |
create_metadata_summary |
Combined metadata file |
|
Creates metadata column summary |
Descriptions of Processes in BIN_ASSESSMENT Workflow#
prepareInputFiles: Prepares input genome bins by standardizing file extensions and handling compressed files.
Input: Directory containing genome bins
Output: Directory with standardized genome bins in
.faformat
runCheckM2: Assesses genome quality using CheckM2, which estimates completeness and contamination based on lineage-specific marker genes.
Input: Standardized genome bins
Output: CheckM2 quality report and protein files
Uses CheckM2 database for marker gene identification
runGUNC: Assesses genome contamination using GUNC, which detects contamination based on genomic consistency.
Input: Protein files from CheckM2
Output: GUNC contamination report
Uses GUNC database for reference-based contamination detection
combineFiles: Combines CheckM2 and GUNC results, calculates quality scores, and filters genomes based on quality thresholds.
Input: CheckM2 and GUNC reports
Output: Combined quality report and filtered genome bins
Implements quality filtering based on the selected quality parameter
gtdbtk: Performs taxonomic classification of quality-filtered genomes using GTDB-Tk, which assigns taxonomy based on the Genome Taxonomy Database.
Input: Quality-filtered genome bins
Output: GTDB-Tk classification results for bacterial and archaeal genomes
Utilizes GTDB-r220 database for classification
createFinalFile: Creates a comprehensive final report combining quality assessment and taxonomic classification.
Input: Combined quality report and GTDB-Tk results
Output: Final report with quality metrics and taxonomy
Merges results from previous runs if available
combineMetadata: (Optional) Combines the final quality/taxonomy report with user-provided metadata.
Input: Final report and user metadata file
Output: Combined metadata file with quality, taxonomy, and sample information
Links genomic information with sample metadata for downstream analysis
create_metadata_summary: Creates a summary of metadata columns to assist in subsequent analyses.
Input: Combined metadata file
Output: Metadata column summary with indices and names
Helps identify relevant columns for downstream analysis
Tools Used in BIN_ASSESSMENT#
Tool |
Purpose |
Version |
Database |
Database Version |
Default Parameters |
Parameters that can be selected |
|---|---|---|---|---|---|---|
CheckM2 |
Genome quality assessment |
1.0.2 |
CheckM2 Database |
1.0.2 |
|
Output directory name |
GUNC |
Genome contamination assessment |
1.0.6 |
GUNC Database |
progenomes2.1 |
|
Output directory name |
GTDB-Tk |
Genome taxonomy classification |
2.4.0 |
GTDB |
r220 |
|
None specific to this workflow |
Custom Scripts in BIN_ASSESSMENT#
Script |
Purpose |
Input |
Output |
Note |
|---|---|---|---|---|
Checkm2_GUNC_combine_quality_pass.py |
Combines CheckM2 and GUNC results |
CheckM2 and GUNC reports |
Combined quality report |
Calculates quality scores and flags |
GTDB_add2_check2gunc.py |
Combines quality and taxonomy data |
Quality report and GTDB-Tk results |
Combined quality/taxonomy report |
Creates comprehensive genome report |
combine_metadata_WMS_genome.py |
Combines genome data with sample metadata |
Genome report and user metadata |
Combined metadata file |
Links genomic data with sample context |
Usage Notes#
The BIN_ASSESSMENT workflow is designed to work with the output from the ASSEMBLY_BINNING workflow.
Providing metadata with the
--metadataoption is highly recommended for downstream analysis in the COMPARATIVE_ANNOTATION module.The default quality filter (
QS50.pass) selects genomes with a QS50 quality score ≥ 50, but you can choose different filters based on your research needs.The script checks for the existence and non-emptiness of the input directory before proceeding.
For parallel processing of multiple samples, you can run the workflow with different
--run_idvalues.The final quality/taxonomy report and optional combined metadata file are key inputs for the COMPARATIVE_ANNOTATION module.
Next Steps#
After assessing genome quality and taxonomy with this module, proceed to GENOME_SELECTOR to select genomes of you interest. Then, use the result csv file in COMPARATIVE_ANNOTATION to:
Perform gene prediction and annotation
Conduct comparative genomic analyses
Generate functional profiles
Compare genomes across different samples or conditions
Combining Metadata with Quality/Taxonomy Results#
Combining Sample Metadata with Genome Information
The metadata should contain a common basename of paired-end read metagenomic files in CSV format.
For example, the base name of a pair of paired-end metagenomic fastq files would be:
SRR6915091_1.fastq, SRR6915091_2fastq –> basename: SRR6915091
Metadata of Metagenomes
Example table content of metadata csv file:
You need to prepare metadata file in csv format of your own.
For your reference, download metadata of bioproject PRJNA447983 in csv format.
CRC_Control113_PRJNA447983_metadata.csv
bioproject_accession |
accession_used_in_analysis |
country |
continent |
host_age |
host_body_mass_index |
host_sex |
disease_group |
AJCC_stage |
age_group |
|---|---|---|---|---|---|---|---|---|---|
PRJNA447983 |
SRR6915092 |
Italy |
Europe |
60 |
20 |
Female |
Control |
Control |
Old |
PRJNA447983 |
SRR6915097 |
Italy |
Europe |
80 |
32 |
Female |
Control |
Control |
Old |
PRJNA447983 |
SRR6915108 |
Italy |
Europe |
67 |
20.43816558 |
Female |
Control |
Control |
Old |
PRJNA447983 |
SRR6915113 |
Italy |
Europe |
77 |
NA |
Female |
Control |
Control |
Old |
In this table, the second column accession_used_in_analysis is the base name of generated MAGs.
Metadata of MAGs
Upon successful execution of the
BIN_ASSESSMENTmodule, thequality_taxonomy_combined_final.csvfile is generated with genome quality, taxonomy, and detailed metrics information:
Genome,Completeness,Contamination,medium_quality.pass,high_quality.pass,medium_quality_gunc.pass,high_quality_gunc.pass,QS50,QS50.pass,pass.GUNC,QS50_gunc.pass,Completeness_Model_Used,Translation_Table_Used,Coding_Density,Contig_N50,Average_Gene_Length,Genome_Size,GC_Content,Total_Coding_Sequences,Total_Contigs,Max_Contig_Length,classification
ERR1018185qced_headed_MB2.1,62.18,0.38,True,False,True,False,60.28,True,True,True,Neural Network (Specific Model),11,0.886,3309,254.5395019981556,2799002,0.51,3253,900,18216,d__Bacteria;p__Pseudomonadota;c__Gammaproteobacteria;o__Enterobacterales;f__Enterobacteriaceae;g__Escherichia;s__Escherichia coli
Combined Metadata File
When using the
--metadataoption with--accession_column, thecombined_metadata_quality_taxonomy_${run_id}.csvfile is generated:
Genome |
Completeness |
Contamination |
medium_quality.pass |
high_quality.pass |
classification |
bioproject_accession |
accession_used_in_analysis |
country |
host_age |
disease_group |
|---|---|---|---|---|---|---|---|---|---|---|
SRR6915091_bin.1 |
91.77 |
0.55 |
True |
True |
d__Bacteria;p__Bacillota_C;c__Negativicutes;o__Acidaminococcales;f__Acidaminococcaceae;g__Acidaminococcus;s__Acidaminococcus intestini |
PRJNA447983 |
SRR6915091 |
Italy |
77 |
CRC |
SRR6915091_bin.2 |
93.32 |
6.58 |
True |
False |
d__Bacteria;p__Bacteroidota;c__Bacteroidia;o__Bacteroidales;f__Bacteroidaceae;g__Phocaeicola;s__Phocaeicola dorei |
PRJNA447983 |
SRR6915091 |
Italy |
77 |
CRC |
This combined file links genome quality and taxonomy with sample metadata, providing valuable context for downstream analyses in the COMPARATIVE_ANNOTATION module.
To generate this combined file, use:
(metafun) metafun -module BIN_ASSESSMENT -m your_metadata.csv -c 2
Where 2 refers to the column index in your metadata that contains the sample identifiers matching your genome bin names.
The output file will be named with a timestamp and workflow identifier (e.g., combined_metadata_quality_taxonomy_20250306135829_2996.csv), where:
20250306135829is the timestamp in format yyyyMMddHHmmss2996is a unique workflow identifier
BIN_ASSESSMENT is a crucial step that bridges the gap between genome recovery and functional analysis, ensuring that only high-quality genomes are used for downstream comparative studies.