metaFun에 오신 것을 환영합니다#
#
metaFun : An analysis pipeline for metagenomic big data with fast and unified Functional searches#
metaFun은 Nextflow와 apptainer로 구현되었습니다. conda 또는 mamba를 사용하여 쉽게 설치하고 실행할 수 있습니다. 이 패키지는 Bioconda 채널(https://anaconda.org/bioconda/metafun)에 등록되어 있습니다.
소개#
metaFun은 메타게놈 어셈블 게놈(MAG)의 빠르고 확장 가능한 생성과 통계 분석을 포함한 분류학적 프로파일링을 목표로 합니다. 사용자가 관심 있는 게놈과 메타데이터를 사용하여 빠른 비교 유전체 분석과 기능 주석을 가능하게 합니다.
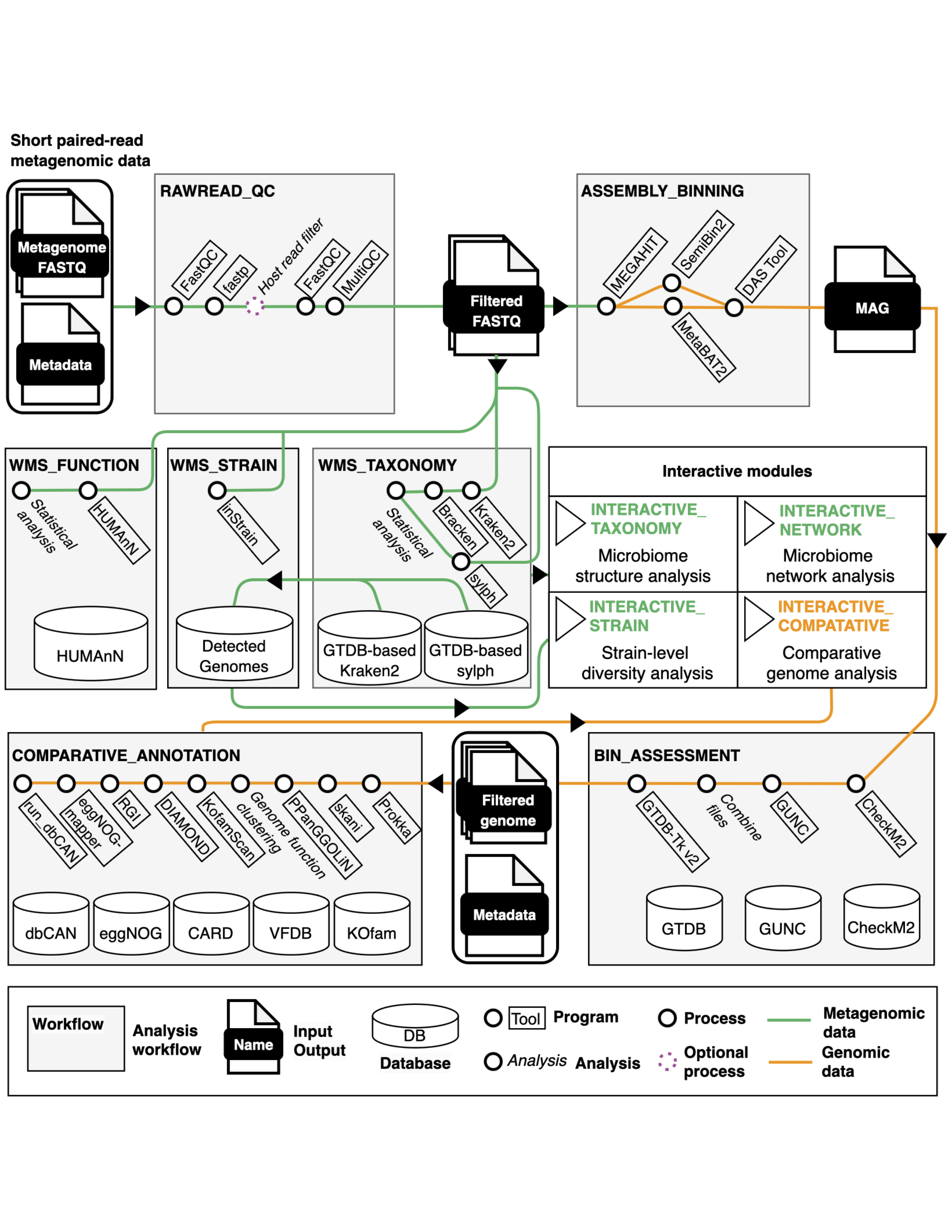
metaFun 파이프라인의 조망도. 이 파이프라인은 일곱 개의 분석 모듈과 네 개의 인터랙티브 모듈로 구성되어 있습니다.#
빠른 시작#
필수 구성 요소 설치 (conda, miniconda, or mamba)
# Linux OS를 사용한다고 가정합니다. wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh -O ~/miniconda3/miniconda.sh # -p $PATH를 교체하여 설치 경로를 지정할 수 있습니다. $PATH는 conda 설치의 기본 디렉토리입니다. bash miniconda.sh -b -u -p ~/miniconda3 rm miniconda.sh
metaFun 설치
# metafun 환경 만들기 conda create -n metafun bioconda::metafun conda activate metafun
데이터베이스 다운로드
(metafun) metafun -module DOWNLOAD_DB # 도움말 보기 (metafun) metafun -help
FAQ 및 문제 해결#
저장 공간 관리#
디스크 사용량 줄이기: 결과를 확인한 후 Nextflow
work/디렉토리를 안전하게 삭제하여 상당한 디스크 공간을 확보할 수 있습니다:# 작업 완료 후 work 디렉토리 제거 rm -rf work/
임시 파일: 여러 모듈은 처리 중에 큰 임시 파일을 생성하며, 이는 성공적인 실행 후 삭제할 수 있습니다:
HUMAnN3 (
WMS_FUNCTION 결과의 *_humann_temp디렉토리)어셈블리 파일 (ASSEMBLY_BINNING 중간 파일)
MetaPhlAn bowtie2 인덱스 (WMS_TAXONOMY에서)
선택적 결과 보존: 대규모 메타제노믹 연구의 경우, 필수 출력만 보관하는 것을 고려하세요:
최종 테이블 및 시각화 파일 저장
gzip으로 큰 텍스트 출력 압축성공적인 빈 정제 후 원시 빈닝 결과 보관
자주 발생하는 문제#
메타데이터 형식: 가장 일반적인 오류는 메타데이터 파일 문제에서 발생합니다:
메타데이터 CSV 파일의 샘플 ID가 읽기 파일 이름의 접두사와 정확히 일치하는지 확인
매개변수에 지정된 열 번호(
-s/-c,-a)가 올바른지 확인CSV 파일이 탭이나 세미콜론이 아닌 쉼표(,) 구분자를 사용하는지 확인
인터랙티브 모듈의 경우, 분석 단계 전반에 걸쳐 일관된 메타데이터 확인
데이터베이스 설치: 데이터베이스 관련 오류가 발생하면 필요한 데이터베이스를 다시 설치해야 할 수 있습니다:
# 특정 데이터베이스 재설치 (metafun) metafun -module DOWNLOAD_DB -d humann3 # WMS_FUNCTION용 (metafun) metafun -module DOWNLOAD_DB -d kraken2 # WMS_TAXONOMY용 # 모든 데이터베이스 완전 재설치 (metafun) metafun -module DOWNLOAD_DB
메모리 요구 사항: 여러 모듈은 상당한 메모리를 필요로 합니다:
ASSEMBLY_BINNING: OOM 오류가 발생하면 metaSPAdes의
-m매개변수를 낮춤WMS_FUNCTION: HUMAnN3의
-p매개변수로 스레드 조정저메모리 환경의 경우, 작은 배치로 샘플 처리
시작하기
- 1. 빠른 시작
- 1.1. metaFun 설치 및 실행
- 1.2. RAWREAD_QC: 원시 리드의 품질 제어 및 숙주 게놈 필터링
- 1.3. ASSEMBLY_BINNING: 어셈블리 및 빈닝
- 1.4. BIN_ASSESSMENT: 게놈 품질 평가 및 분류학적 분류
- 1.5. GENOME_SELECTOR: 게놈 선택 인터페이스
- 1.6. COMPARATIVE_ANNOTATION: 비교 게놈 분석
- 1.7. INTERACTIVE_COMPARATIVE: 대화형 비교 분석
- 1.8. WMS_TAXONOMY: 메타게놈 리드의 분류학적 프로파일링
- 1.9. INTERACTIVE_TAXONOMY: 대화형 분류학 분석
- 1.10. WMS_FUNCTION: 메타게놈 리드의 기능 분석
- 1.11. WMS_STRAIN: 균주 수준 미세다양성 분석
- 1.12. INTERACTIVE_STRAIN: 대화형 균주 다양성 분석
- 1.13. INTERACTIVE_NETWORK: 대화형 미생물 네트워크 분석
- 1.14. DOWNLOAD_DB: 필요한 데이터베이스 다운로드
- 1.15. PREPARE_CUSTOM_HOST: 사용자 정의 숙주 게놈 인덱스 준비
- 1.16. 공통 옵션
metaFun 워크플로우