ASSEMBLY_BINNING#
 This module is a part of metaFun pipeline, designed for de novo assembly, binning, and binning refinement of metagenomic data.
This module is a part of metaFun pipeline, designed for de novo assembly, binning, and binning refinement of metagenomic data.
Overview#
The ASSEMBLY_BINNING module is for de novo assembly and binning and refinement process. It performs de novo assembly of quality-controlled metagenomic reads, followed by metagenomic binning to recover metagenome-assembled genomes (MAGs) with refinement process.
Module Execution#
# Basic usage
(metafun) metafun -module ASSEMBLY_BINNING
# Specify input directory if you used a custom output path in RAWREAD_QC
(metafun) metafun -module ASSEMBLY_BINNING -i /path/to/filtered_reads
# Change MEGAHIT assembly parameters
(metafun) metafun -module ASSEMBLY_BINNING --megahit_presets meta-large
# Use SemiBin2 in self-training mode (learn features from your input data, takes longer time)
(metafun) metafun -module ASSEMBLY_BINNING --semibin2_mode self
# Use specific environment model for SemiBin2 (several environment models are available)
# 'human_gut','dog_gut','ocean','soil','cat_gut','human_oral','mouse_gut','pig_gut','built_environment','wastewater','chicken_caecum','global'
(metafun) metafun -module ASSEMBLY_BINNING --semibin2_mode human_gut
Assembly and binning options
There are several options to optimize your assembly and binning process: You can specify all options in one command line.
MEGAHIT assembly presets:
Default value is set to
default, which is balanced for most metagenomes.For large and complex metagenomes such as soil , use
--megahit_presets meta-large:
(metafun) metafun -module ASSEMBLY_BINNING --megahit_presets meta-large
For more sensitive but slower assembly, use
--megahit_presets meta-sensitive:
(metafun) metafun -module ASSEMBLY_BINNING --megahit_presets meta-sensitive
SemiBin2 environment models:
Default value is set to
self(self-supervised learning without reference models).For novel environments without reference data, use the self-supervised mode:
(metafun) metafun -module ASSEMBLY_BINNING --semibin2_mode self
For specific environments, choose from the available models:
# Human microbiome
(metafun) metafun -module ASSEMBLY_BINNING --semibin2_mode human_gut
(metafun) metafun -module ASSEMBLY_BINNING --semibin2_mode human_oral
# Animal microbiomes
(metafun) metafun -module ASSEMBLY_BINNING --semibin2_mode dog_gut
(metafun) metafun -module ASSEMBLY_BINNING --semibin2_mode cat_gut
(metafun) metafun -module ASSEMBLY_BINNING --semibin2_mode mouse_gut
(metafun) metafun -module ASSEMBLY_BINNING --semibin2_mode pig_gut
(metafun) metafun -module ASSEMBLY_BINNING --semibin2_mode chicken_caecum
# Environmental samples
(metafun) metafun -module ASSEMBLY_BINNING --semibin2_mode ocean
(metafun) metafun -module ASSEMBLY_BINNING --semibin2_mode soil
(metafun) metafun -module ASSEMBLY_BINNING --semibin2_mode wastewater
(metafun) metafun -module ASSEMBLY_BINNING --semibin2_mode built_environment
# General purpose model
(metafun) metafun -module ASSEMBLY_BINNING --semibin2_mode global
Module Operation Sequence#
This module performs the following steps:
De novo assembly using MEGAHIT
Contig renaming for consistent format
Building Bowtie2 index for assembled contigs
Metagenomic read mapping to contigs
Metagenomic binning using MetaBAT2 and SemiBin2
Bin refinement using DAS Tool
Parameters#
${launchDir} is the directory where you execute metaFun, and utilized as output base directory.
Parameter |
Description |
Default Value |
Note |
|---|---|---|---|
|
Input directory with filtered reads |
|
Output from RAWREAD_QC module or specify your own directory containing quality-filtered reads. You can run without input directory if you did not specify output directory in RAWREAD_QC module. |
|
Output directory |
|
Default recommended for downstream analysis |
|
MEGAHIT assembly preset |
|
Options: |
|
SemiBin2 environment model |
|
Options: |
|
Number of CPUs to use |
|
Inputs and Outputs#
Inputs#
Quality-controlled paired-end metagenomic reads (from RAWREAD_QC workflow)
These reads should be host-filtered and quality-trimmed
Default input directory:
${launchDir}/results/metagenome/RAWREAD_QC/read_filtered
Outputs#
Assembled contigs (FASTA format)
Metagenome-assembled genomes (MAGs) from multiple binning methods:
MetaBAT2 bins
SemiBin2 bins
DAS Tool refined bins using MetaBAT2 and SemiBin2
Associated mapping files and intermediate results (can be assessed in work directory of Nextflow)
Output directory structure#
Output directory is at ${launchDir}/results/metagenome/ASSEMBLY_BINNING or your specified directory path with -o outdir.
Switching input and output directory.
If you define a custom output directory with -o ${output directory}, you should modify input parameters in downstream workflows accordingly.
The default output directory is results/metagenome/ASSEMBLY_BINNING in your ${launchDir}.
${launchDir}/results/metagenome/ASSEMBLY_BINNING/
├── assembled_contigs/ # MEGAHIT assembly results
│ ├── results/
│ │ ├── assembly/
│ │ │ ├── ${sample_id}/
│ │ │ │ ├── ${sample_id}_renamed_MH.contigs.fa # Assembled and renamed contigs
│ ├── ...
├── metabat2_bins/ # MetaBAT2 binning results
│ ├── ${sample_id}_MB2.1.fa # Individual MetaBAT2 bin
│ ├── ${sample_id}_MB2.2.fa
│ ├── ${sample_id}_MB2.3.fa
│ ├── ...
├── semibin2_bins/ # SemiBin2 binning results
│ ├── ${sample_id}_${mode}_SB2_0.fa # Individual SemiBin2 bin
│ │ # where ${mode} is the selected environment model (e.g., self, human_gut)
│ ├── ${sample_id}_${mode}_SB2_1.fa
│ ├── ${sample_id}_${mode}_SB2_2.fa
│ ├── ...
├── dastool_bins/ # DAS Tool refined bins
│ ├── ${sample_id}_dastool_DASTool_bins/
│ │ ├── ${sample_id}_*.fa # DAS Tool refined bin
│ │ ├── ...
│ ├── ...
└── final_bins/ # Final collection of all bins for downstream analysis
├── ${sample_id}_*.fa # Copies of the best bins from all methods
├── ...
Execution Examples and Results#
metaFun command line execution example#
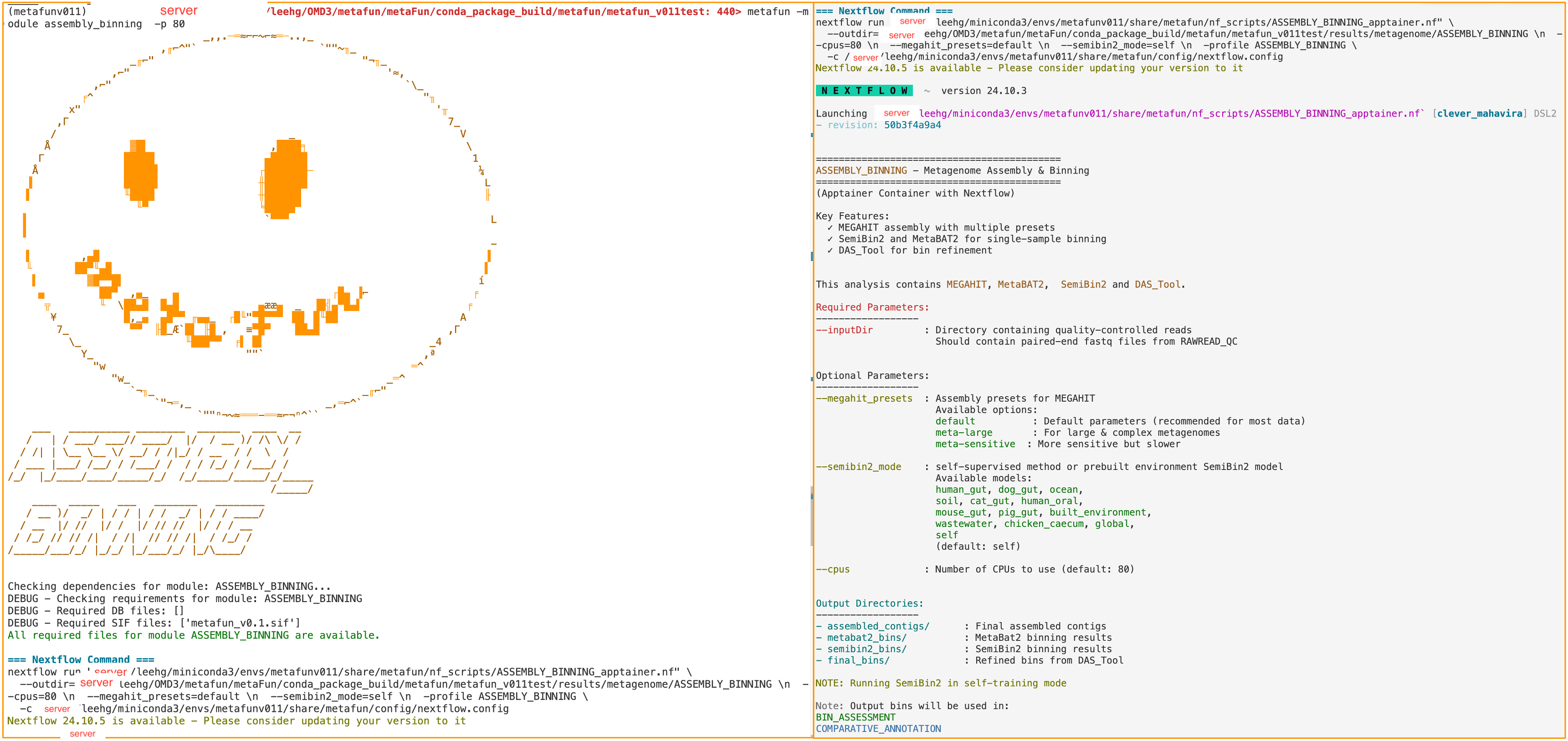
About SemiBin2 output file naming
The SemiBin2 output file names follow the pattern: ${sample_id}_${mode}_SB2_${number}.fa
${sample_id}: The sample identifier from your input reads${mode}: The SemiBin2 environment model you selected with--semibin2_modeExample:
self,human_gut,ocean, etc.
${number}: Sequential bin number
Example: SRR6915091_human_gut_SB2_14.fa
Assemblis in assmebled_contigs folder and genomes in the final_bins directory are the main output files of this module.
The quality of generated bins can be assessed in BIN_ASSESSMENT module.
Nextflow Processes in ASSEMBLY_BINNING Module#
Process |
InputDir |
OutputDir |
Note |
|---|---|---|---|
AssemblyAndRename |
|
|
MEGAHIT assembly and contig renaming |
Bowtie2IndexBuild |
Output from AssemblyAndRename |
Intermediate result |
Builds Bowtie2 index for contigs |
MHcontig2sortedbam |
Reads and Bowtie2 index |
Intermediate result |
Maps reads to contigs and creates sorted BAM |
MB2_binning |
Sorted BAM and contigs |
|
MetaBAT2 binning |
SB2_binning |
Sorted BAM and contigs |
|
SemiBin2 binning |
Contigs2bin_prep_mb2 |
MetaBAT2 bins |
Intermediate result |
Prepares MetaBAT2 bin info for DAS Tool |
Contigs2bin_prep_sb2 |
SemiBin2 bins |
Intermediate result |
Prepares SemiBin2 bin info for DAS Tool |
Dastool |
MetaBAT2 and SemiBin2 bin info, contigs |
|
DAS Tool bin refinement |
get_bins |
All bins from DAS Tool |
|
Collects all bins for downstream analysis |
Descriptions of Processes in ASSEMBLY_BINNING Workflow#
AssemblyAndRename: Performs de novo assembly using MEGAHIT and renames contigs for consistent format. Creates output in
assembled_contigs/results/assembly/${sample_id}/${sample_id}_renamed_MH.contigs.fa.Bowtie2IndexBuild: Builds Bowtie2 index for the assembled contigs to facilitate read mapping. Generates index files named
${sample_id}_MH_bt2_index.*.bt2that are used in the mapping step.MHcontig2sortedbam: Maps reads to contigs using Bowtie2 and creates sorted BAM files for binning. Produces
${sample_id}_sorted.bamand${sample_id}_sorted.bam.baifiles needed for both binning methods.MB2_binning: Performs metagenomic binning using MetaBAT2, which bins contigs based on coverage and sequence composition. Generates bins with naming pattern
${sample_id}_MB2.${number}.fa.SB2_binning: Performs metagenomic binning using SemiBin2, which uses deep learning for binning. Creates bins with naming pattern
${sample_id}_${mode}_SB2_${number}.fa, where${mode}is the selected environment model.Contigs2bin_prep_mb2 and Contigs2bin_prep_sb2: Prepare contig-to-bin files for DAS Tool integration. These processes generate the mapping files between contigs and bins required by DAS Tool.
Dastool: Refines and integrates bins from both binning methods to produce higher quality MAGs. Outputs refined bins in
${sample_id}_dastool_DASTool_bins/directory.get_bins: Collects all generated bins and places them in the
final_bins/directory for easy access in downstream analysis. This directory serves as the primary input for the BIN_ASSESSMENT module.
Tools Used in ASSEMBLY_BINNING#
Tool |
Purpose |
Version |
Default parameters |
Parameters that can be selected |
|---|---|---|---|---|
MEGAHIT |
De novo assembly |
1.2.9 |
Varies based on |
|
Bowtie2 |
Read mapping |
2.5.2 |
|
Not specified in this script |
MetaBAT2 |
Metagenomic binning |
2.15 |
|
Not specified in this script |
SemiBin2 |
Metagenomic binning |
2.1.0 |
|
|
DAS Tool |
Bin refinement |
1.1.7 |
|
Not specified in this script |
Usage Notes#
The script checks for the existence and non-emptiness of the input directory before proceeding.
SemiBin2 can be run in self-supervised mode or with premade environment models by setting
--semibin2_mode ${mode}:Use
selffor novel environments without reference genomesAvailable environment models:
human_gut,dog_gut,ocean,soil,cat_gut,human_oral,mouse_gut,pig_gut,built_environment,wastewater,chicken_caecum,global
For complex metagenomes, consider using
--megahit_presets meta-largefor better assemblyFor sensitive assembly (more compute-intensive), use
--megahit_presets meta-sensitiveThe ASSEMBLY_BINNING workflow is designed to work with the output from the RAWREAD_QC workflow.
The resulting bins can be assessed for quality and taxonomic classification in the BIN_ASSESSMENT module.
Next Steps#
After generating MAGs with this module, proceed to BIN_ASSESSMENT to:
Assess genome quality using CheckM2 and GUNC
Classify taxonomy using GTDB-Tk
Filter bins based on quality metrics
Combine results with metadata for downstream analysis