INTERACTIVE_STRAIN#
 이 모듈은 InStrain에서 생성된 균주 수준 다양성 결과를 탐색하고 분석하기 위한 대화형 웹 인터페이스를 제공하는 metaFun 파이프라인의 일부입니다.
이 모듈은 InStrain에서 생성된 균주 수준 다양성 결과를 탐색하고 분석하기 위한 대화형 웹 인터페이스를 제공하는 metaFun 파이프라인의 일부입니다.
개요#
INTERACTIVE_STRAIN 모듈은 균주 수준 메타게놈 데이터를 시각화하고 분석하기 위한 동적 웹 기반 플랫폼을 제공합니다. 연구자들이 뉴클레오타이드 다양성 패턴을 대화형으로 탐색하고, pN/pS 비율을 통해 선택압을 분석하며, 샘플 간 공유 균주를 식별하고, 다양성 지표와 메타데이터 변수 간의 상관관계를 분석할 수 있습니다. 이 모듈은 전처리된 InStrain 출력을 활용하여 프로그래밍 지식 없이도 고급 통계 분석과 맞춤형 시각화를 가능하게 합니다.
이 모듈은 명령줄 분석의 대화형 대안을 제공하여, 사용자가 사전에 지정하지 않고도 다양한 지표와 비교를 동적으로 탐색할 수 있습니다.
모듈 실행#
# WMS_STRAIN 결과를 사용한 기본 사용법
(metafun) metafun -module INTERACTIVE_STRAIN -i results/metagenome/WMS_STRAIN
# 웹 인터페이스용 사용자 정의 포트 지정
(metafun) metafun -module INTERACTIVE_STRAIN -i results/metagenome/WMS_STRAIN -p 8080
# 추가 메타데이터 파일 추가
(metafun) metafun -module INTERACTIVE_STRAIN -i results/metagenome/WMS_STRAIN -m updated_metadata.csv
모듈 작동 순서#
이 모듈은 다음 단계를 수행합니다:
균주 수준 데이터 로딩 (WMS_STRAIN 결과로부터):
모든 샘플의 InStrain 프로파일 출력 읽기
뉴클레오타이드 다양성 및 pN/pS 데이터 통합
게놈 주석을 위한 GTDB 분류 메타데이터 로딩
샘플 메타데이터와 결합
대화형 웹 서버 실행 (여러 분석 모듈 포함):
균주 분포 시각화를 위한 공유 분류군 분석
미세다양성 분석을 위한 뉴클레오타이드 다양성 탐색기
선택압 조사를 위한 pN/pS 분석
통합 분석을 위한 다양성-pN/pS 상관관계
대화형 구성요소를 통한 온디맨드 분석 활성화:
게놈, 샘플 또는 메타데이터별 동적 필터링
조건 간 통계 테스트
맞춤형 시각화 옵션
다운스트림 응용을 위한 데이터 내보내기
매개변수#
매개변수 |
설명 |
기본값 |
참고 |
|---|---|---|---|
|
WMS_STRAIN 결과가 있는 입력 디렉토리 |
필수 |
InStrain 프로파일을 포함한 WMS_STRAIN 출력 경로 |
|
추가 메타데이터 파일 |
선택 사항 |
업데이트되거나 추가된 샘플 메타데이터가 있는 CSV/TSV 파일 |
|
GTDB 메타데이터 파일 |
자동 감지 |
게놈 분류 정보가 있는 TSV 파일 |
|
웹 인터페이스용 포트 번호 |
|
기본 포트가 이미 사용 중이면 조정 |
|
사용할 CPU 코어 수 |
|
계산 집약적 작업용 |
입력 및 출력#
입력#
완료된 WMS_STRAIN 실행의 결과 디렉토리 (InStrain 프로파일 출력 포함)
향상된 분석을 위한 CSV/TSV 형식의 선택적 추가 메타데이터 파일
분류 주석을 위한 선택적 GTDB 메타데이터 파일
출력#
다양한 형식의 내보낸 시각화 (PNG, PDF, SVG)
통계 분석 결과 (CSV, TSV)
필터링된 데이터 테이블
출판 준비 그림
출력 디렉토리 구조#
생성된 파일은 타임스탬프가 있는 출력 디렉토리에 저장됩니다:
${launchDir}/results/interactive_strain/YYYYMMDDHHMMSS/
├── exported_figures/ # 내보낸 시각화
│ ├── shared_taxa_venn_[timestamp].pdf # Venn 다이어그램
│ ├── shared_taxa_upset_[timestamp].pdf # UpSet 플롯
│ ├── nucleotide_diversity_[timestamp].pdf # 다양성 박스플롯/히트맵
│ ├── pnps_analysis_[timestamp].pdf # pN/pS 분포 플롯
│ └── correlation_plot_[timestamp].pdf # 다양성 vs pN/pS 상관관계
├── statistical_results/ # 통계 테스트 결과
│ ├── diversity_stats_[timestamp].csv # 다양성 통계
│ ├── pnps_comparison_[timestamp].csv # pN/pS 비교 결과
│ └── correlation_results_[timestamp].csv # 상관관계 분석
└── exported_data/ # 내보낸 데이터 테이블
├── shared_taxa_matrix_[timestamp].csv # 분류군 공유 행렬
├── diversity_data_[timestamp].csv # 뉴클레오타이드 다양성 값
└── pnps_data_[timestamp].csv # pN/pS 비율 데이터
인터페이스 구성요소#
웹 인터페이스는 여러 탭으로 나뉘며, 각 탭은 다양한 유형의 균주 수준 분석을 위한 전문 도구를 제공합니다.
메인 인터페이스 구조#
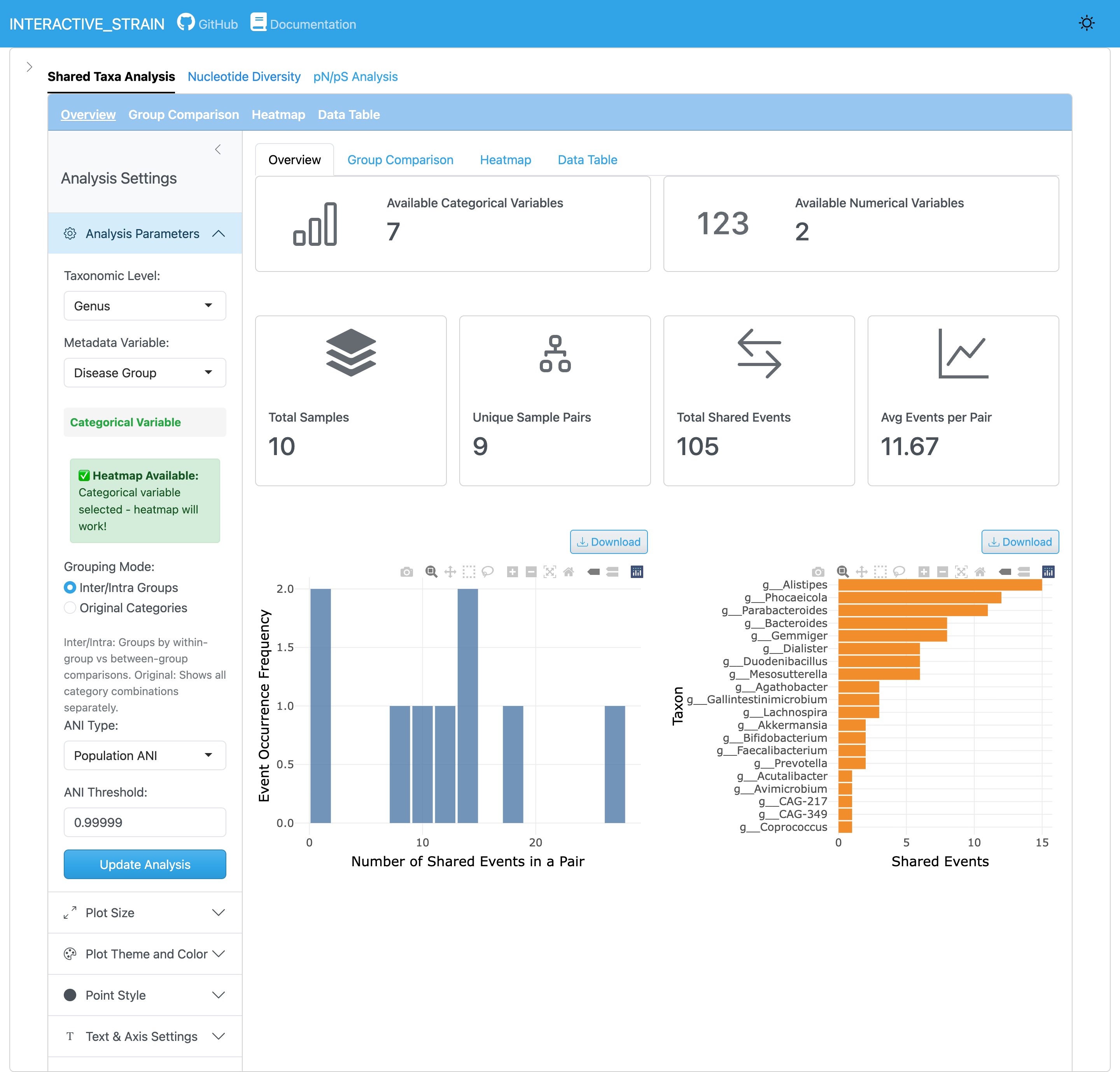
메인 인터페이스는 매개변수 구성을 위한 사이드바 패널, 다양한 분석 모듈에 접근하기 위한 탐색 탭(공유 분류군 분석, 뉴클레오타이드 다양성, pN/pS 분석, 다양성 vs pN/pS), 그리고 분석 결과를 표시하는 메인 콘텐츠 영역으로 구성됩니다.
사이드바 패널#

사이드바 패널은 WMS_STRAIN 결과를 로드하기 위한 컨트롤을 제공합니다. 사용자가 InStrain 출력 디렉토리 경로를 지정하면 애플리케이션이 integrated_microbiome_data.rds 파일에서 샘플 메타데이터를 포함한 모든 프로파일 데이터를 자동으로 로드합니다. 선택적 입력에는 분류 주석을 위한 GTDB 메타데이터 파일과 필요시 추가 샘플 메타데이터 파일이 포함됩니다.
popANI와 conANI 이해하기
InStrain은 균주 비교를 위해 두 가지 ANI 지표를 계산합니다:
popANI (집단 수준 ANI): 주요 및 소수 대립유전자를 모두 고려하여 샘플 내 미세다양성을 설명
conANI (합의 ANI): 샘플 간 합의(주요) 대립유전자만 비교
공유 균주 식별에는 99.999% popANI 기본 임계값이 권장됩니다. 사용자는 연구 필요에 따라 공유 분류군 분석 모듈에서 이러한 임계값을 조정할 수 있습니다. 자세한 내용은 InStrain 문서를 참조하세요.
탭별 분석 설계
이 인터페이스의 모든 분석은 해당 탭 내에서만 적용되도록 설계되었습니다. 각 분석 모듈은 독립적으로 작동하며:
한 탭에서 변경된 설정은 다른 탭의 분석에 영향을 미치지 않음
각 탭은 자체 상태 및 구성을 유지
한 탭에서 생성된 결과는 해당 탭의 분석에만 특정
이러한 모듈식 설계로 간섭 없이 다른 매개변수로 다양한 분석을 동시에 실행 가능
공유 분류군 분석#
이 모듈은 샘플 간 공유된 균주/게놈을 식별하고 시각화합니다.
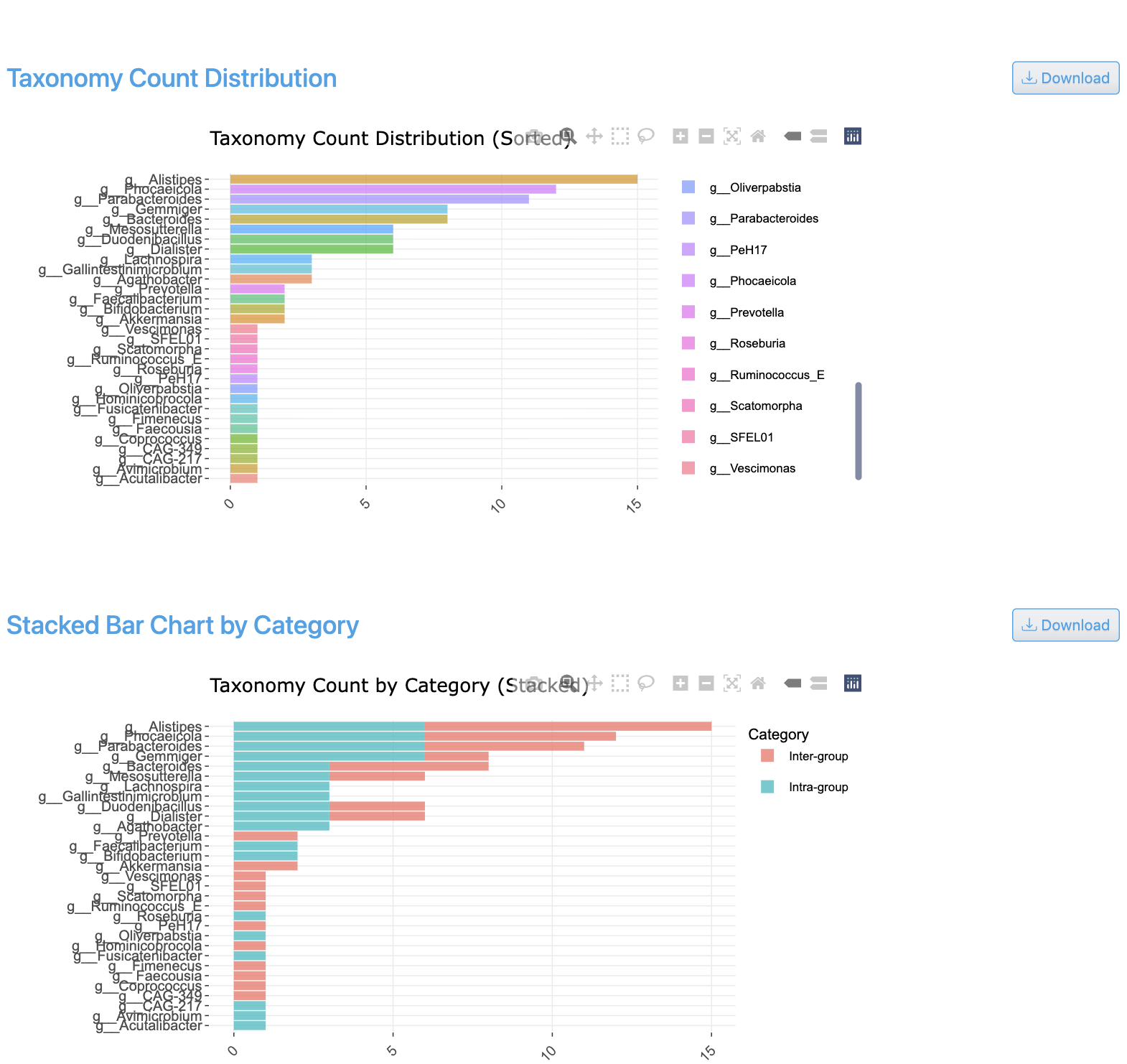
공유 분류군 카운트 뷰는 샘플 그룹 간 겹치는 분류군을 보여주는 Venn 다이어그램과 UpSet 플롯을 표시합니다. 조건 간 공유 및 고유 게놈 수를 시각화하며, 모든 그룹 조합에 대한 교집합 크기를 제공합니다.
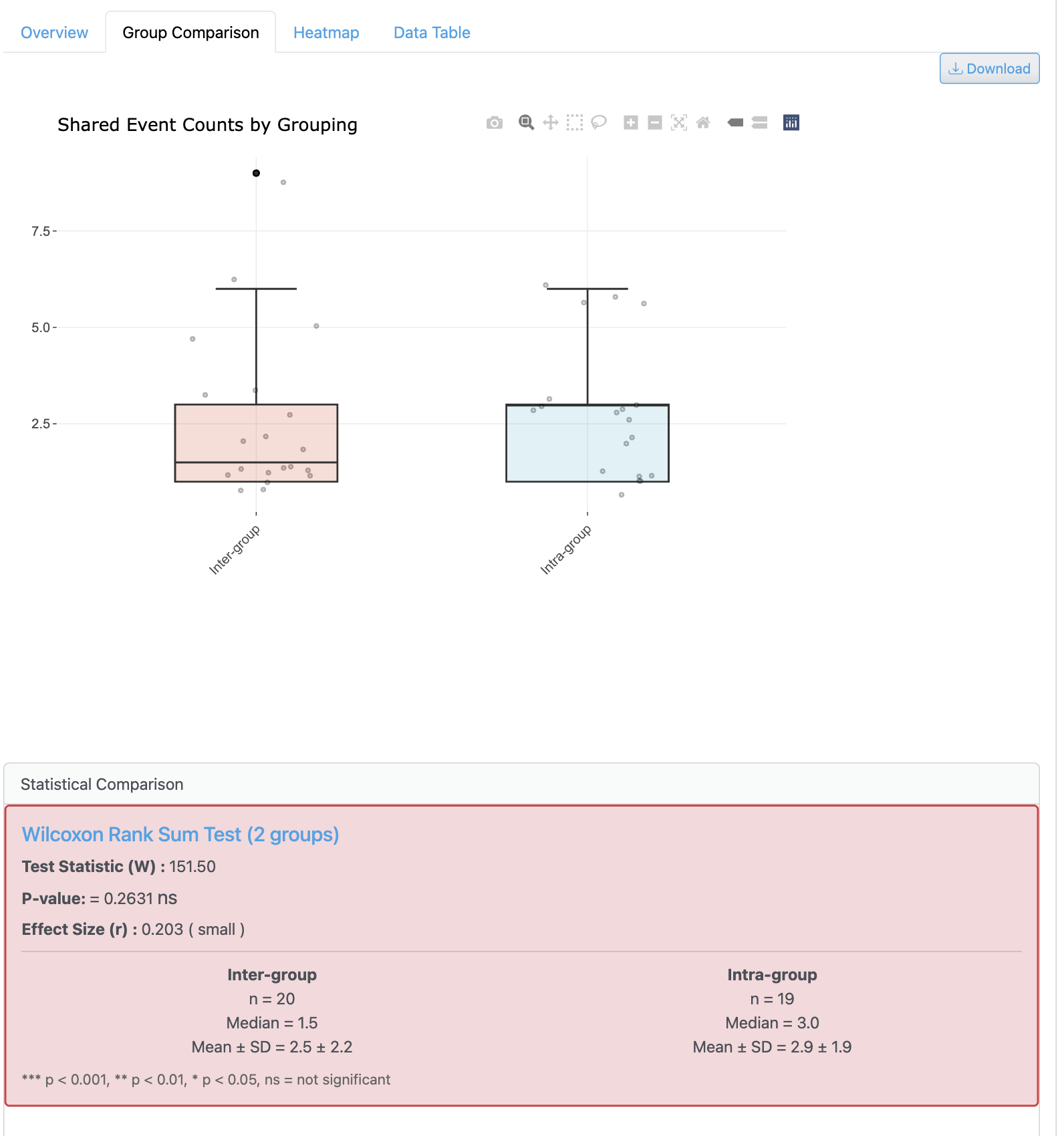
그룹 기반 공유 분석은 메타데이터 카테고리 간 균주 분포 패턴 비교를 가능하게 합니다. 통계 요약과 함께 다양한 실험 조건 간 게놈이 어떻게 분포되어 있는지 보여줍니다.
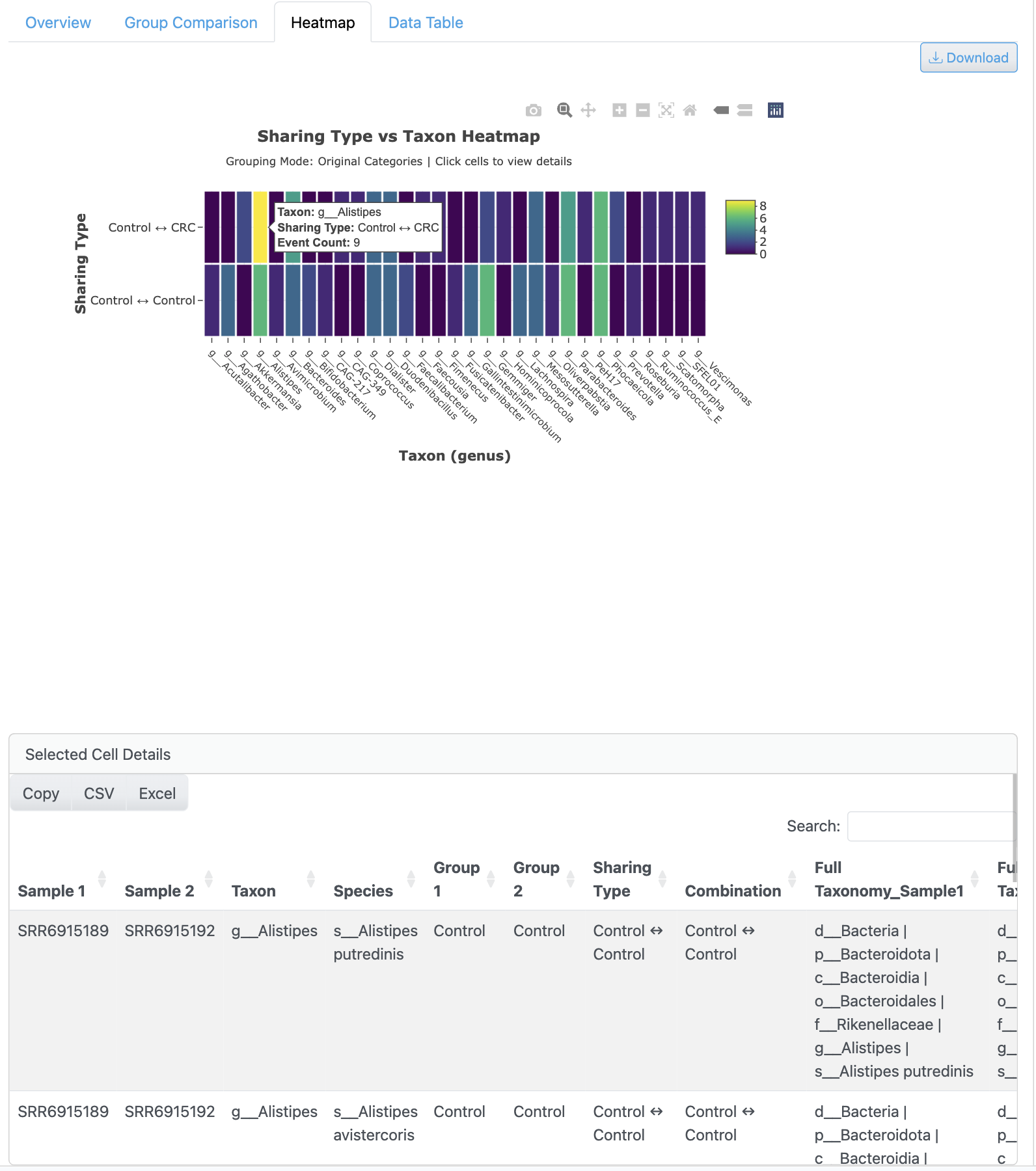
존재/부재 히트맵은 모든 샘플에 걸친 게놈 검출의 행렬 시각화를 제공합니다. 계층적 클러스터링은 샘플 그룹화 패턴을 보여주며, 검출 상태를 나타내는 색상 코딩과 prevalence 필터링 옵션이 있습니다.
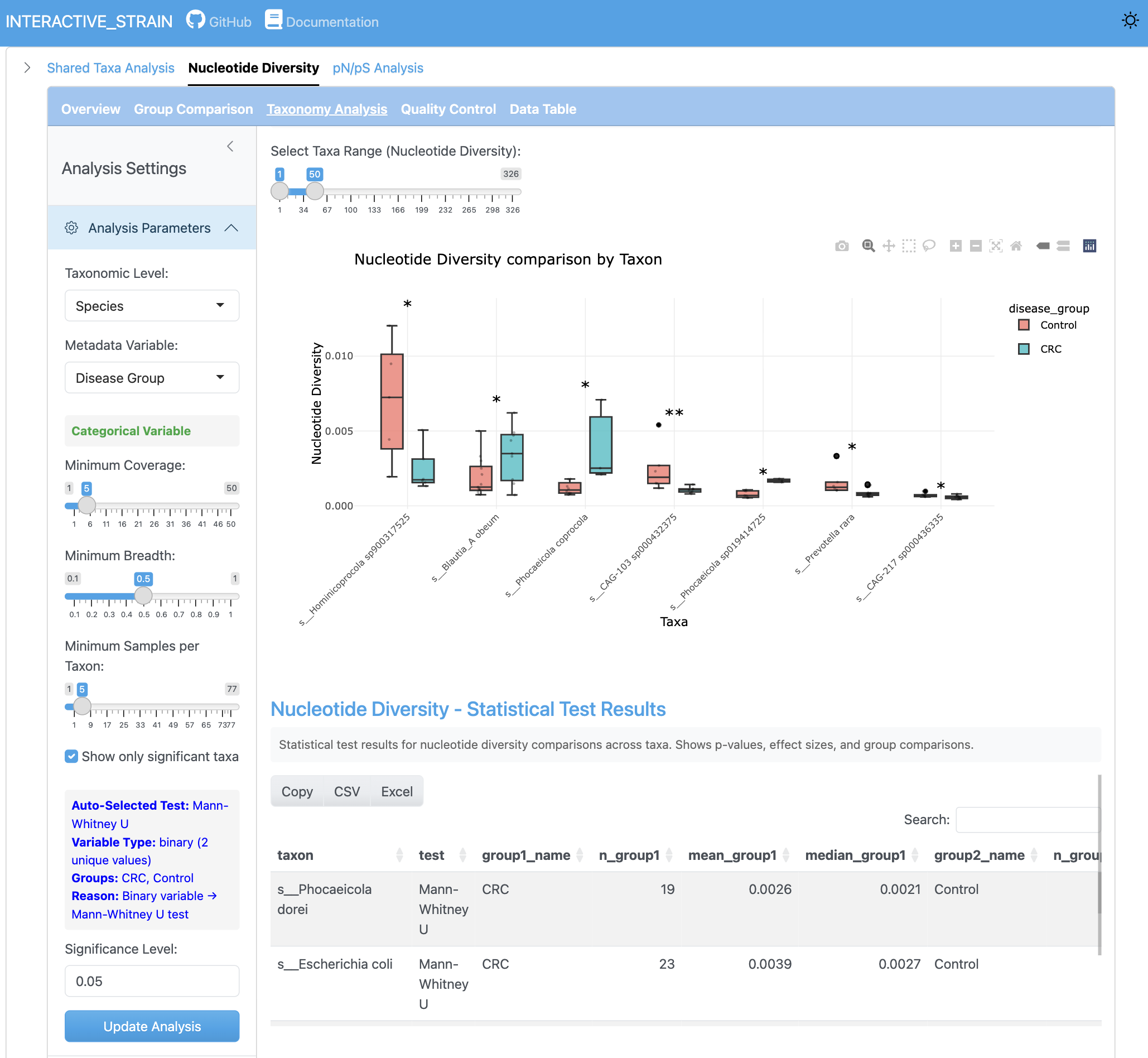
뉴클레오타이드 다양성 탐색기#
집단 내 유전적 다양성의 종합 분석입니다.
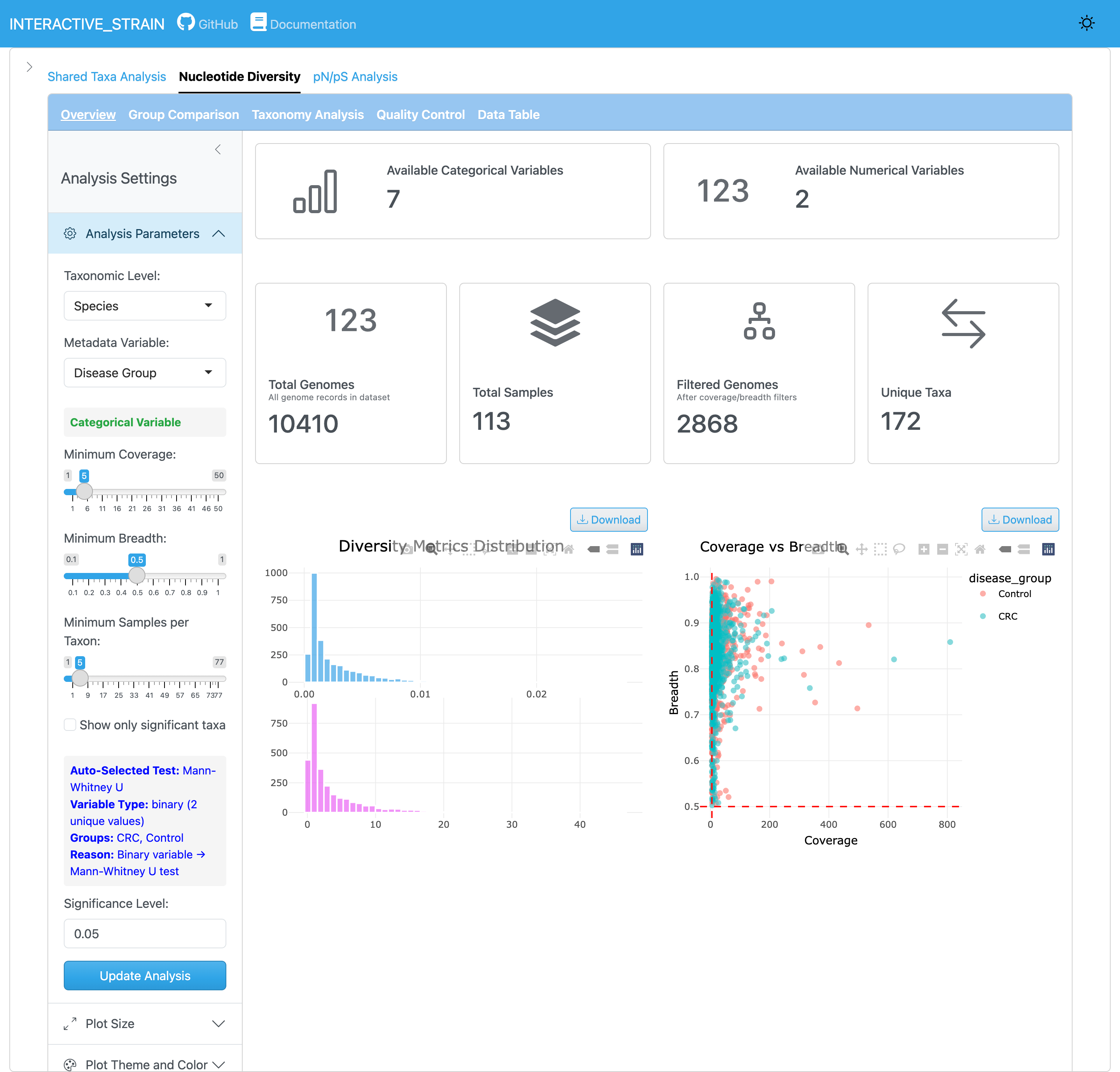
개요 패널은 모든 게놈과 샘플에 걸친 뉴클레오타이드 다양성(π) 값의 분포를 표시합니다. InStrain은 Nei and Li (1979) 방법을 사용하여 뉴클레오타이드 다양성을 계산합니다: π = 1 - Σ(각 염기의 빈도)², 충분한 커버리지(기본값 ≥5x)를 가진 모든 게놈 위치에서 계산하고 유전자/게놈 전체의 평균을 구합니다. 이 지표는 집단 내 유전적 변이를 정량화하며 샘플 간 커버리지 차이에 강건합니다. 패널은 신뢰성 있는 미세다양성 추정을 위한 요약 통계와 품질 필터링 옵션을 제공합니다.
다양성 분포 뷰는 히스토그램과 밀도 플롯으로 상세한 뉴클레오타이드 다양성 패턴을 보여줍니다. 사용자는 다양성 값의 범위를 탐색하고 높거나 낮은 미세다양성을 가진 게놈을 식별할 수 있습니다.
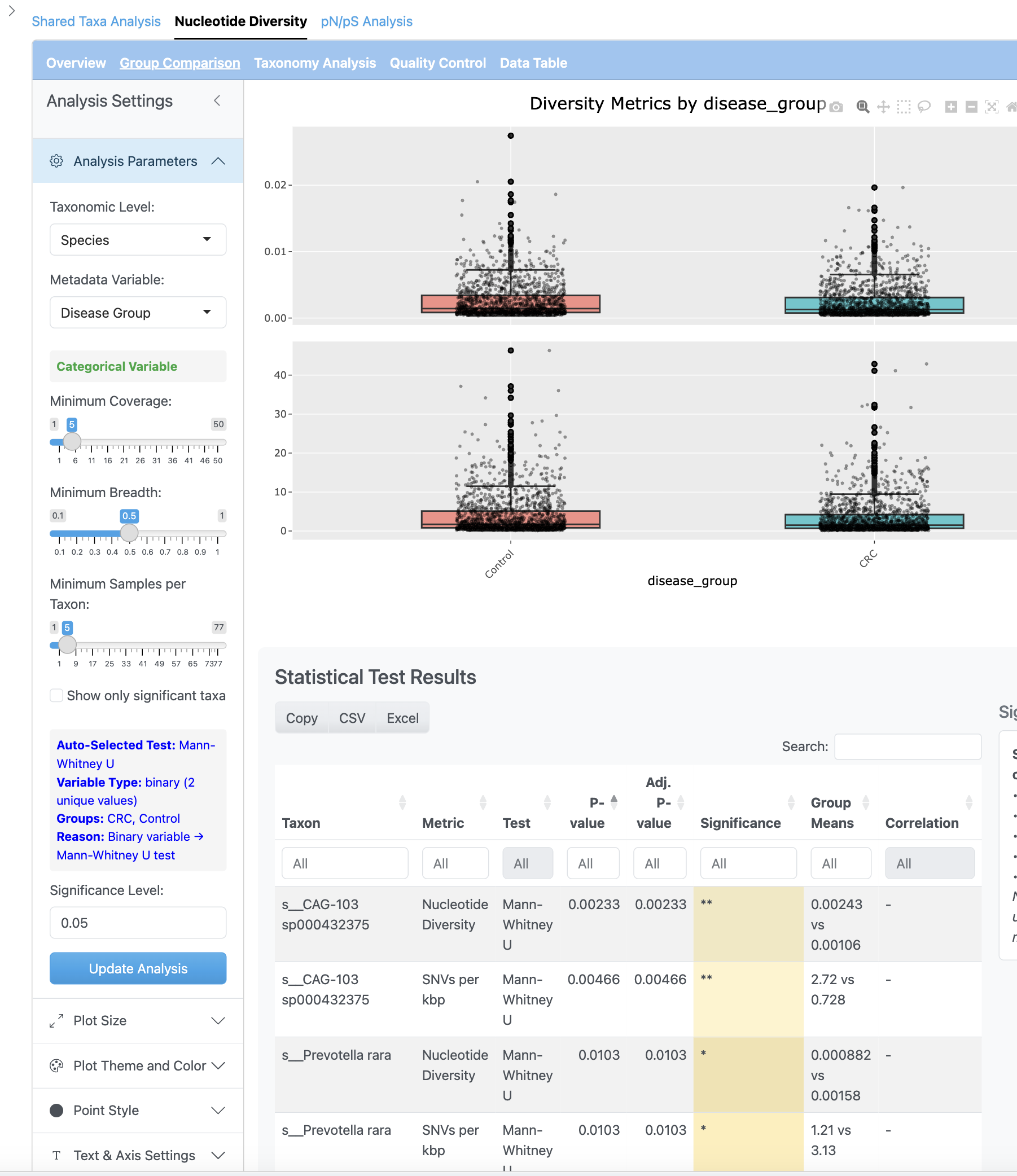
그룹 비교 뷰는 메타데이터 그룹 간 뉴클레오타이드 다양성을 비교하는 박스플롯을 제시합니다. 효과 크기 계산과 다중 검정 보정 옵션과 함께 통계 테스트(Wilcoxon, Kruskal-Wallis)가 수행됩니다.
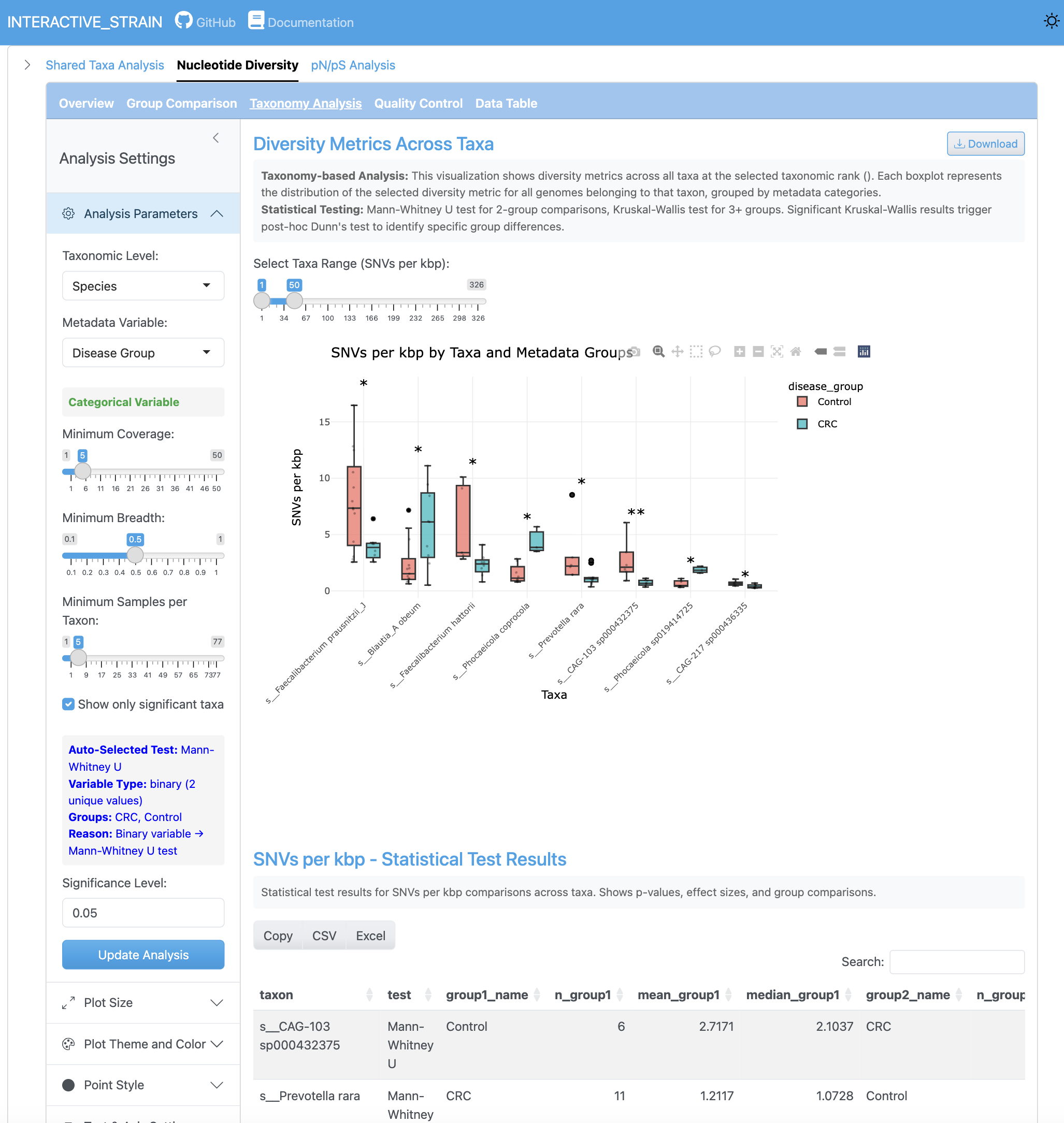
SNV 밀도 분석은 게놈과 샘플 전체에 걸친 킬로베이스 쌍당 단일 염기 변이 수를 보여줍니다. 이 지표는 돌연변이율과 집단 수준의 유전적 변이에 대한 통찰을 제공합니다.
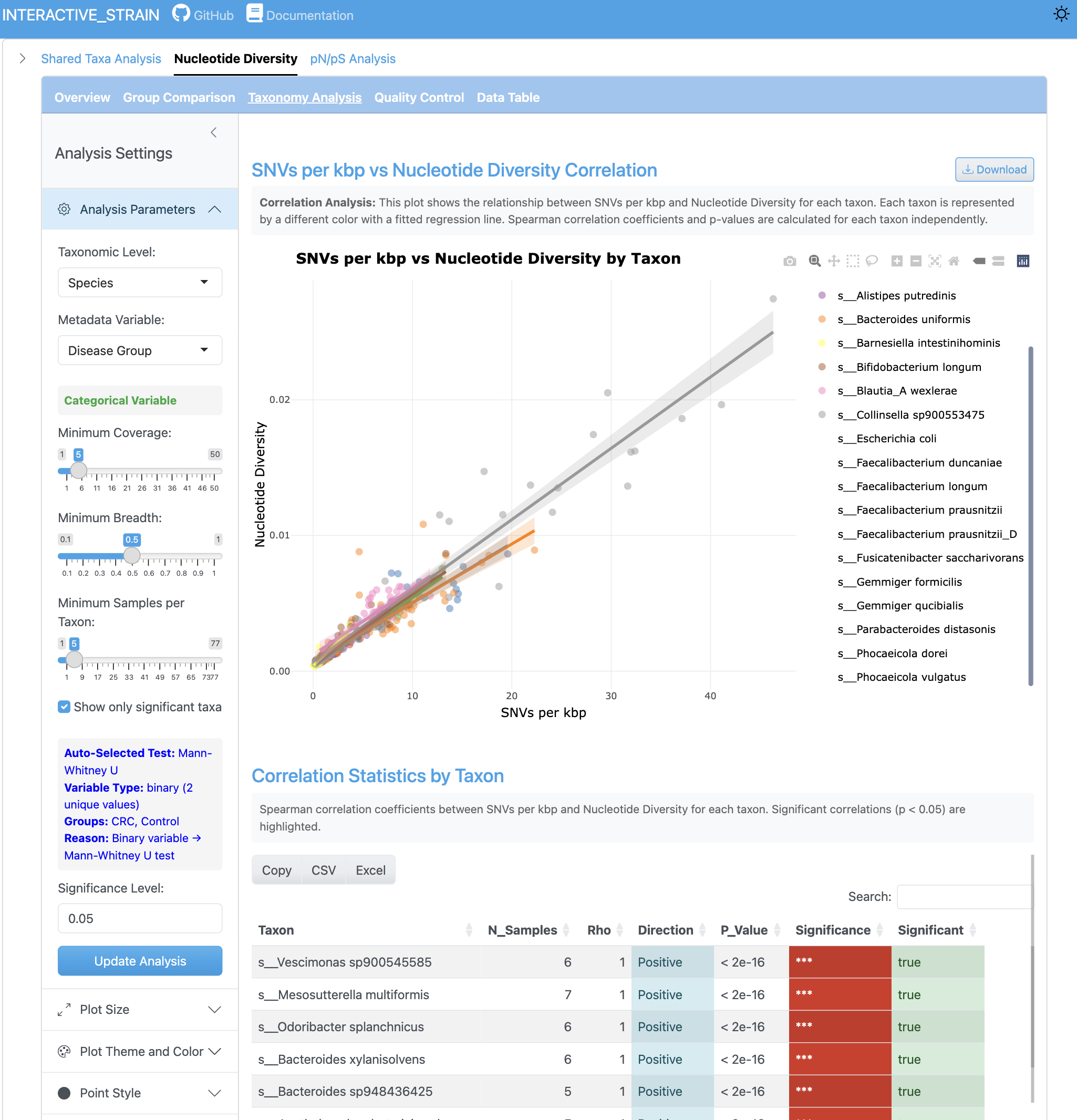
상관관계 뷰는 SNV 카운트와 뉴클레오타이드 다양성 지표 간의 관계를 표시합니다. 회귀선이 있는 산점도는 다양한 게놈과 샘플 조건에서 이러한 지표들이 어떻게 관련되는지 보여줍니다.
pN/pS 분석#
여러 수준에서의 선택압 분석입니다.
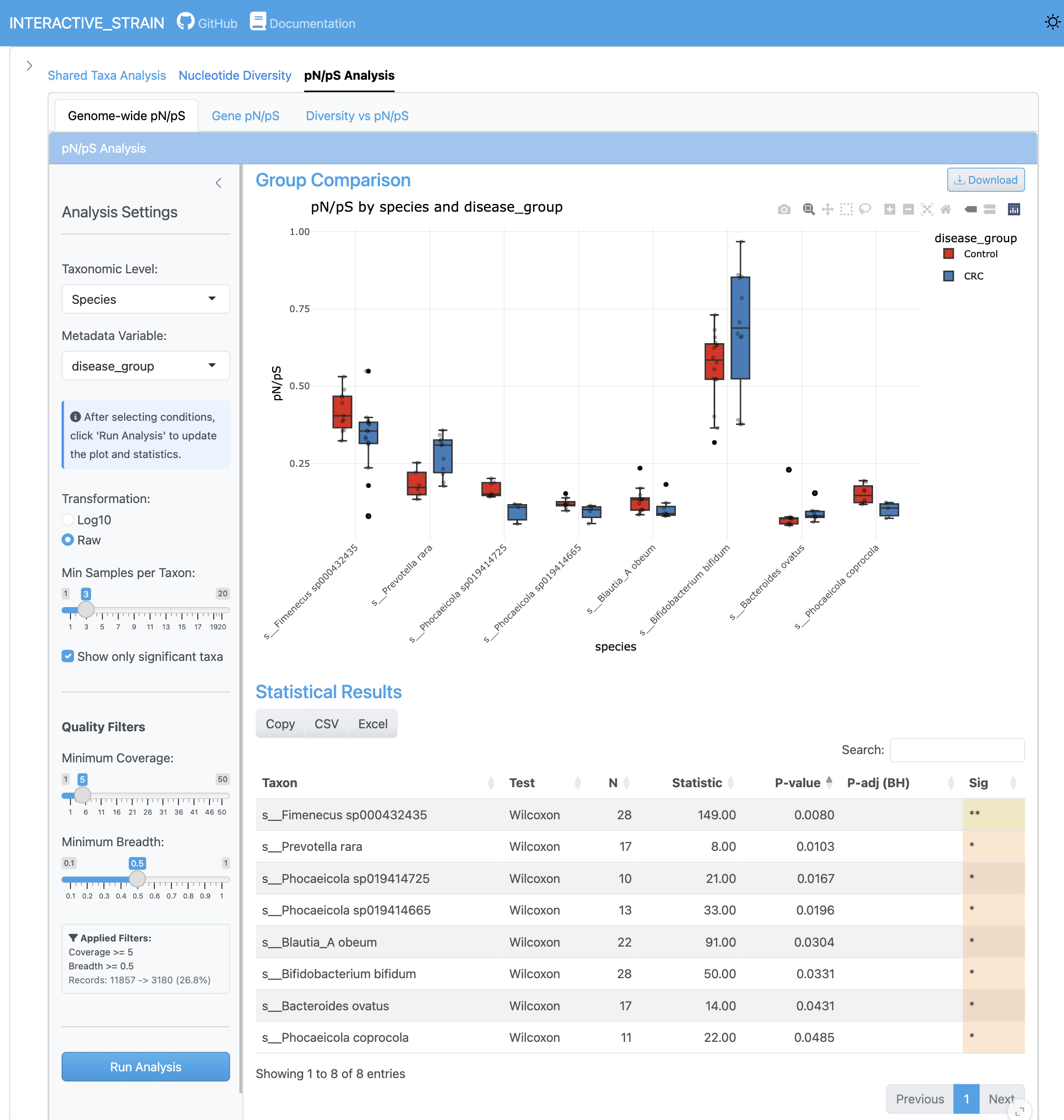
게놈 전체 pN/pS 분석은 모든 게놈에 걸친 pN/pS 비율의 분포를 표시하여 선택압을 받는 게놈을 식별합니다. 통계적 유의성 테스트가 있는 그룹 비교는 조건 간 차별적 선택 패턴을 보여줍니다.
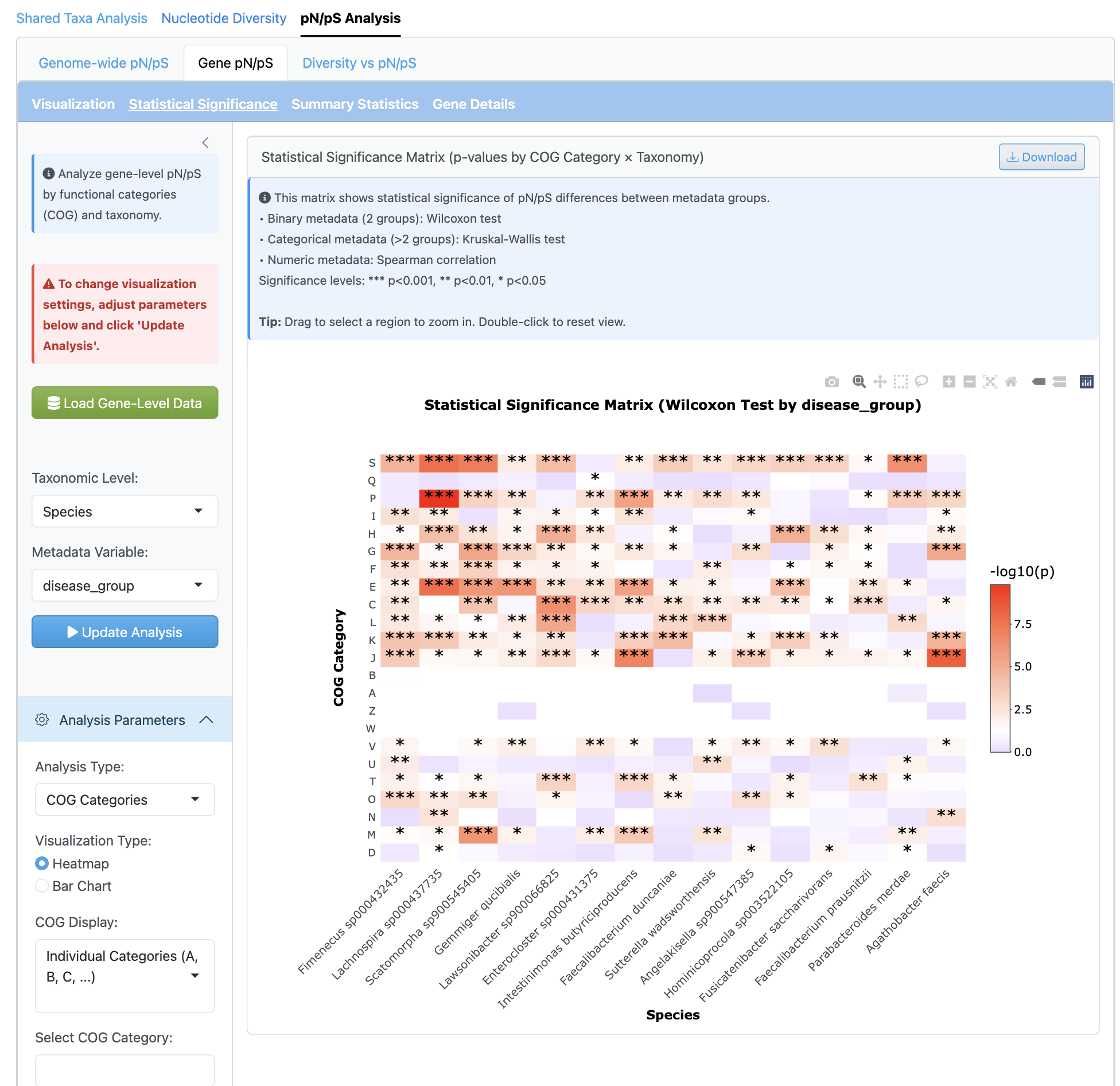
유전자 수준 분석은 기능 주석 통합(COG 카테고리)과 함께 유전자별 세밀한 선택압 조사를 제공합니다. 기능 카테고리별 히트맵 시각화는 양성 또는 정화 선택을 받는 유전자 식별을 가능하게 합니다.
다양성 vs pN/pS 상관관계#
다양성과 선택의 통합 분석입니다.
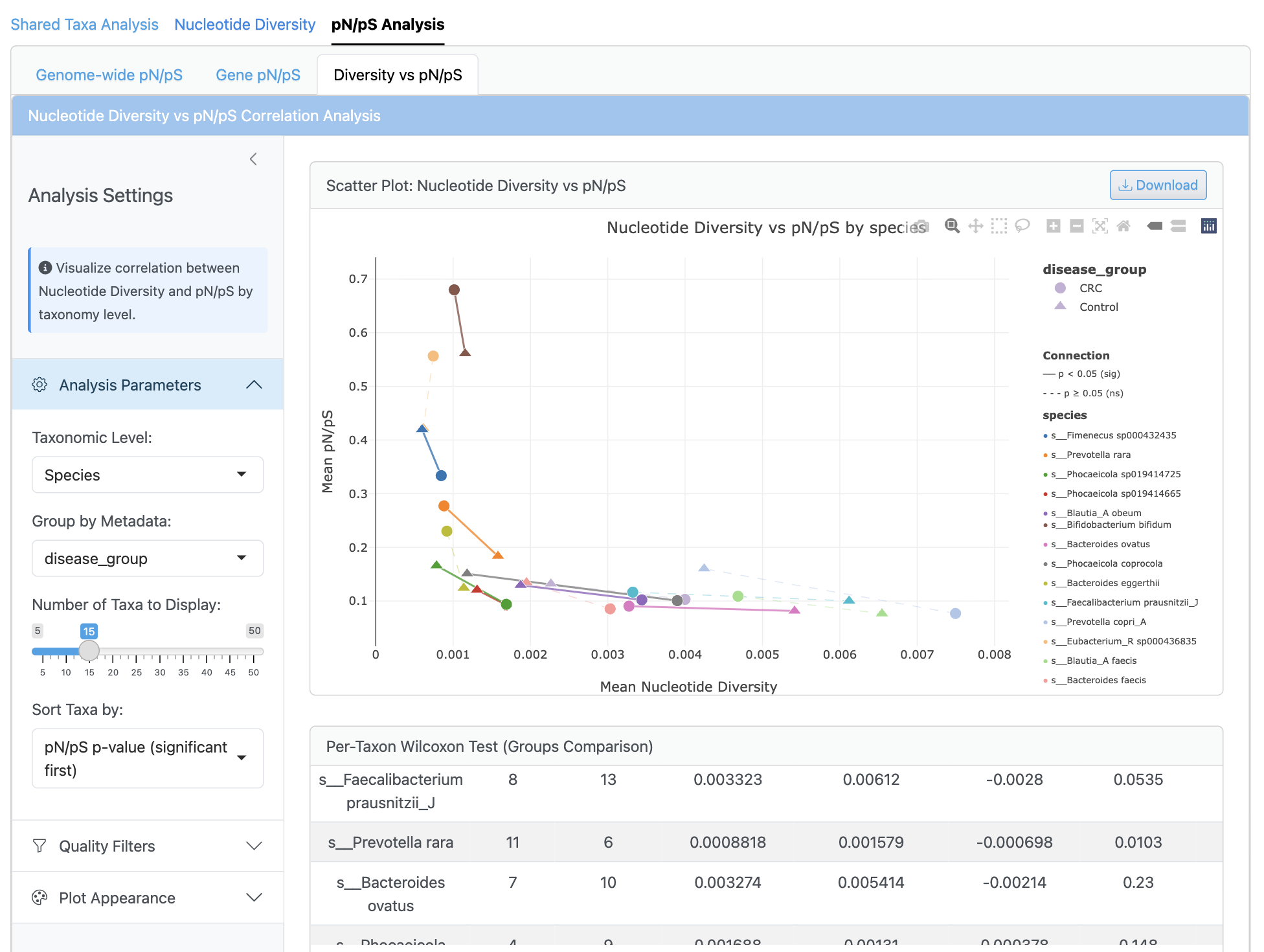
상관관계 분석은 회귀선과 신뢰 구간이 있는 뉴클레오타이드 다양성 대 pN/pS 비율의 산점도를 표시합니다. 게놈별 및 샘플별 상관관계는 Pearson 및 Spearman 방법을 사용하여 계산되며, 그룹별 분석과 다중 검정 보정이 포함됩니다. 이 통합 뷰는 다양한 조건에서 집단 다양성과 선택압 간의 관계를 보여줍니다.
사용 워크플로우#
INTERACTIVE_STRAIN 모듈의 일반적인 분석 워크플로우:
데이터 입력 패널을 사용하여 InStrain 데이터 로드
공유 분류군 분석을 사용하여 게놈 분포 이해
샘플과 그룹 전체에 걸친 뉴클레오타이드 다양성 패턴 탐색
pN/pS 비율을 사용한 선택압 분석
다양성-선택 관계 조사
출판 준비 그림과 테이블로 결과 내보내기
최적 성능을 위한 팁
대용량 데이터셋의 경우, 반응성 향상을 위해 관심 게놈으로 필터링
높은 신뢰도 데이터에 집중하기 위해 커버리지 임계값 사용
pN/pS 값 해석 시 생물학적 관련성 고려
종합적인 문서화를 위해 시각화와 원시 데이터 모두 내보내기
균주 수준 결과 해석
균주 수준 데이터 분석 시 고려 사항:
커버리지 효과: 저커버리지 게놈은 신뢰할 수 없는 다양성 추정치를 가질 수 있음
샘플 크기: 통계 비교에는 그룹당 적절한 샘플이 필요
생물학적 맥락: pN/pS 해석은 세대 시간과 집단 크기에 따라 다름
다중 게놈: 많은 게놈 분석 시 다중 검정에 주의
사용 참고 사항#
INTERACTIVE_STRAIN 모듈은 WMS_STRAIN 모듈의 InStrain 출력과 함께 작동합니다.
대용량 데이터셋(>50 샘플, >100 게놈)의 최적 성능을 위해 CPU 할당 증가를 고려하세요.
인터페이스가 사용 가능한 게놈과 메타데이터 변수를 자동으로 감지합니다.
웹 인터페이스 사용 중 터미널을 계속 실행하세요; 닫으면 서버가 종료됩니다.
GTDB 메타데이터는 게놈에 대한 분류학적 맥락을 제공하여 분석을 향상시킵니다.
뉴클레오타이드 다양성과 pN/pS 계산에는 충분한 커버리지(일반적으로 >5x)가 필요합니다.
기술 구현#
INTERACTIVE_STRAIN 모듈은 Shiny 프레임워크를 사용하여 구축되었으며 모듈식 아키텍처를 구현합니다:
핵심 구성요소:
app.R: UI 구조와 서버 로직을 정의하는 메인 애플리케이션 파일
helper/plot_customization.R: 플롯 스타일링을 위한 공유 함수
helper/error_handlers.R: 오류 처리 및 사용자 피드백
분석 모듈:
shared_taxa_module.R: 분류군 공유를 위한 Venn 다이어그램과 UpSet 플롯
nucleotide_diversity_module.R: 다양성 분석 및 시각화
pnps_module_quick.R: 게놈 전체 pN/pS 분석
gene_level_pnps_module.R: 유전자 수준 선택 분석
diversity_pnps_correlation_module.R: 통합 상관관계 분석
각 모듈은 자체 UI와 서버 로직을 가진 독립적 구성요소로 설계되어, 애플리케이션 전체에서 일관된 데이터 처리를 보장하면서 독립적인 개발과 유지보수가 가능합니다.
다음 단계#
INTERACTIVE_STRAIN 모듈에서 균주 수준 데이터를 탐색한 후:
WMS_TAXONOMY 모듈을 사용한 분류학적 분석으로 보완
WMS_FUNCTION 모듈로 기능적 잠재력 탐색
INTERACTIVE_NETWORK 모듈을 사용한 미생물 상호작용 조사
R 또는 Python에서 맞춤 분석을 위한 처리된 데이터 내보내기
INTERACTIVE_STRAIN 모듈은 종합적인 균주 수준 분석을 제공하여, 연구자들이 미생물 군집 내 미세다양성 패턴, 선택압 및 진화 역학을 이해할 수 있게 합니다.